

《1、 引言》
1、 引言
自弗里德里希·沃勒(FriedrichWöhler)1828年开创性地合成尿素以来,人们已经开发并提出了越来越复杂的有机化合物全合成的技术[1],来应对越来越多样化和复杂的分子靶标带来的挑战。全合成为天然产品的仿制、验证、修改和反制以及药物的设计和合成做出了重大贡献[2],不仅使科学界而且使社会受益。例如,新型先导化合物的发现是药物设计过程的核心。基于配体和基于结构的虚拟筛选技术是最常用的程序,通过对现有化合物库中的化合物进行快速的片上评估,确定最有可能与蛋白质靶点结合的先导化合物。然而,虚拟筛选技术有两个缺点。第一,确定的化合物已经存在,为新专利留下的空间很小;第二,考虑到与所有可能的化合物的理论空间相比,现有库中探索的化学结构空间非常有限,因此从现有库中检索新的化合物越来越具有挑战性。这些限制导致了新药设计研究的激增,新药设计直接产生新的分子结构以匹配所需的药理特性。虽然从头设计带来的好处是容易为生成的化合物申请专利和探索整个化学空间,其中包括大约1060~10 100个化合物[3],但合成从头设计的分子(通常是不存在的)则是主要障碍。事实上,对于通过虚拟筛选发现的化合物来说,合成的可及性(即化合物合成的难易程度)可能是一个关键问题;也就是说,一个被发现的化合物如果在现成的目录中找不到,也买不到,就需要进行合成。
1969年由Corey和Wipke [4]首次正式提出的回溯合成预测,掀起了一场全合成的革命,提高了合理性和系统性。它设计了一连串的反应,将目标化合物递归分解成更简单的构件,直到达到商业上可获得的起始分子。逆合成预测框架包括两个基本任务:单步逆合成预测和多步逆合成预测。单步逆合成预测的主要目标是预测给定目标分子的可能反应物,如图1所示。通过断开连接,即打破目标分子的原子之间的键,可以得到一个或多个合成物。应该注意的是,作为一个片段的合成物(例如,图1中的亲电“PhC=O+”基团)是一个离子或自由基,通常不作为稳定的物种存在,而与合成物相对应的反应物(如苯甲酰溴)是实际用于合成的化学品。一旦准确和递归的单步逆向合成预测完成后,多步逆向合成预测的重点是规划最佳的反应顺序,使合成步骤的数量、起始分子的成本、产生的废物等达到最小。
《图1》
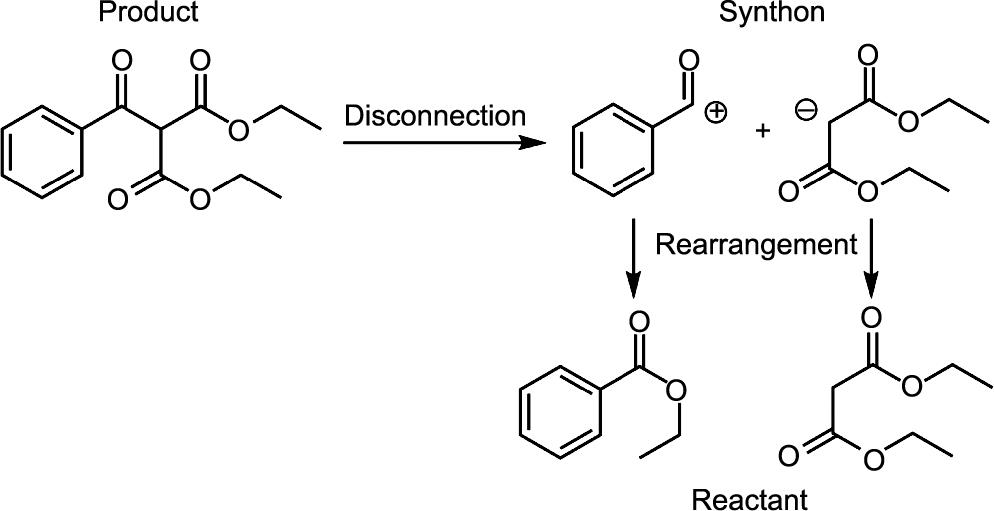
图1 一个单步逆合成反应的例子说明。通过打破给定产品中的一个键,就会产生两个合成物,其中左边的合成物带正电荷,右边的合成物带负电荷。在向真实物质重新排列后,得到了两个反应物。
逆合成预测问题特别具有挑战性。首先,可能的化学反应的数量是巨大的,这就会带来一个巨大的搜索空间。因此,需要有经验的化学家的广泛创造力和专业知识。其次,即使是人类专家,目前对反应机理的理解也是不完整的,在众多可能的逆向合成路线中选择全局最优路线是不容易的。自20世纪60年代以来,人们对协助有经验的化学家进行逆向合成设计的计算机算法给予了很大的关注和期望。基于规则的专家系统[5‒6]是多年来最广泛研究的方法。基于规则的专家系统的核心在于使用逆向合成模板。如图2所示,每个模板由分子子图样表示,编码化学反应期间原子连接的变化。从一个目标分子开始,按照预定的规则选择一个模板,并应用于目标分子以确定反应物。
《图2》
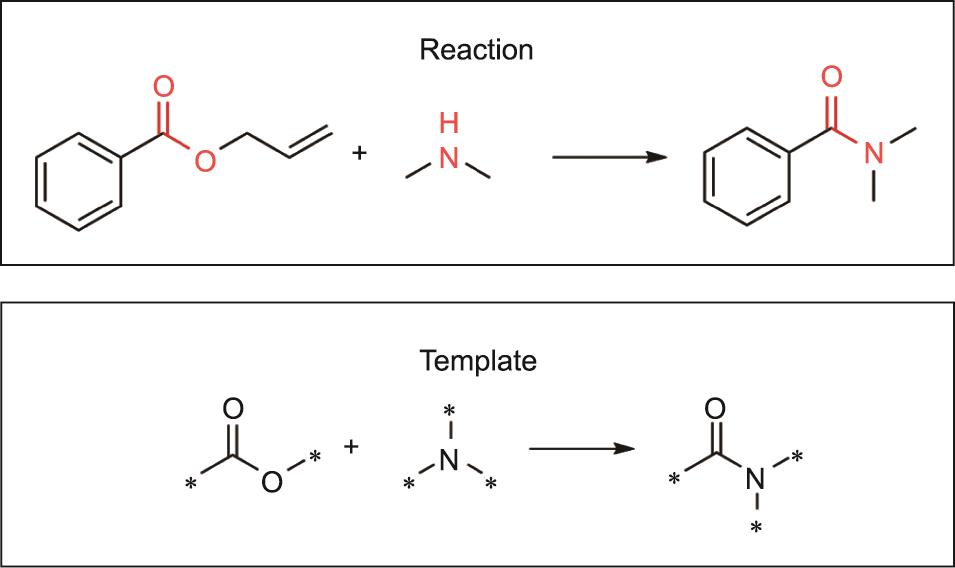
图2 一个化学反应及其逆合成模板的说明。突出显示的部分代表化学反应的反应中心;反应中心的分子子图式被用来组成相应的模板。标有星星的原子是分子中模板外的连接原子。
遗憾的是,基于规则的专家系统存在一些缺点。对这类系统的主要批评是它们没有能力从知识库中为新的目标分子或反应类型提供准确的预测[7‒8]。另一个限制是它们的可扩展性有限。应用成百上千的模板,可以看作是重复解决子图同构问题[9],其计算成本很高,这在许多工作中已经指出[10‒11]。此外,手动编码规则是很费力的[6,12],尽管这些程序如果有足够的时间投入,就可以识别复杂的路线[13]。
最近,人工智能(AI)的广泛成功引发了人们对使用机器学习技术来克服基于规则的专家系统的限制的兴趣。这里,关键的原则是让机器从大型实验数据集中学习化学,以预测反应和进行逆向合成。迄今为止,一系列广泛的研究表明,人工智能辅助模型[13‒15]与基于规则的系统[5‒6]相比,可以实现有竞争力的甚至是更高的逆合成计划性能。在本文的剩余部分,我们将分别讨论现有的用于单步和多步逆合成预测的人工智能算法。
《2、 化学逆合成问题》
2、 化学逆合成问题
《2.1 问题定义》
2.1 问题定义
在进行综述之前,我们首先介绍逆向合成预测问题所涉及的正式定义和记号。
(1)分子。一个含有
(2)化学反应。化学反应是从一组分子到另一组的转化,即
(3)单步逆合成预测。单步逆合成预测问题是化学反应的逆向,它预测了一组反应物ℛ
(4)反应中心。反应中心包括直接参与反应中键和电子重排的原子和键[18]。反应中心对一个化学反应来说是最重要的,因为给定一个具体的反应中心,反应物的预测就容易得多。
(5)合成子。断开产物
(6)模板。一个逆合成反应模板
(1)
式中,
(7)多步逆合成路线规划。给定一个目标产物分子
《2.2 研究挑战》
2.2 研究挑战
图3展示了三条示例性的逆向合成路线,其中前两条是成功的,而最后一条是失败的。为了保证逆向合成路线的准确性和质量,我们在下文中强调了逆向合成规划的关键研究挑战。
《图3》

图3 使用逆向合成计划的两条成功路线和一条失败路线的演示。应该注意的是,所有的路线都是从起始材料开始,以目标分子结束。如果所涉及的每个反应在化学上都是正确的,那么化学家就会评价一条路线是成功的;否则,该路线就是失败的。在本例中,路线失败是因为红色矩形中的反应是不可能的。A、B、C指的是路线中合成的分子。
《2.2.1. 单步逆合成反应预测》
2.2.1. 单步逆合成反应预测
尽管在单步逆合成预测中只生成一步反应,但仍存在几个挑战。根据第2.1节提供的单步逆合成预测的定义,机器会遇到两个主要问题:①如何确定产物的反应中心
同样,另一个“如何生成反应物和试剂”的问题涉及确定哪些反应物和试剂是反应所必需的。一旦反应中心被确定,一个目标分子
《图4》

图4 单步逆合成预测中的三种错误类型。(a)一个错误的分子,其中一个氟原子有三个键;(b)一个错误的反应中心,其中一个溴原子被错误地连接到酯基的邻位;(c)一个不遵循原子守恒的反应,因为一个额外的碳原子被添加到产物中,但在反应物中不存在。
《2.2.2. 多步逆合成路线规划》
2.2.2. 多步逆合成路线规划
由于逆向合成途径的长度可以达到60步或更多[21],逆向合成规划的最终目标是生成一个多步逆向合成途径,这可以通过使用上述一步逆向合成预测算法作为基础来实现。虽然反映反应可行性的单步逆合成预测已被广泛研究并不断改进,但在解决极富挑战性的多步逆合成预测问题上,即以商业化原料库
《3、 相关AI技术综述》
3、 相关AI技术综述
《3.1 序列到序列模型》
3.1 序列到序列模型
由于分子可以直观地表示为一个序列,序列到序列的学习可以作为逆合成预测的有效工具,例如,从一个产品中生成一个反应物的序列。序列到序列学习是一种机器学习方法,已被广泛应用于自然语言处理(NLP)相关的挑战,如语言翻译、图像标题和文本总结。参考文献[23‒24]首次提出了机器翻译问题的编码器-解码器架构:编码器将整个源句子编码为一个固定长度的向量,解码器按顺序生成目标词。Sutskever等[23]和Cho等[24]分别使用递归神经网络(RNN)进行编码和解码。考虑到基于RNN的模型不能捕捉长距离的依赖性,Bahdanau等[25]在基于双向长短时记忆(biLSTM)的框架中引入了注意机制[26],这使得隐藏状态可以考虑全局的上下文信息。Gehring等[27]提出了卷积序列到序列(ConvS2S)模型,该模型使用多层卷积神经网络进行编码和解码,并应用多步注意机制对每个解码层进行全局注意建模。Vaswani等[28]提出了Transformer模型,该模型依靠多头注意力进行编码和解码,使该模型能够在恒定的操作中对输入句子中任意距离的词建立依赖关系。由于变换器模型的优越性能,基于变换器的方法已成为主流,此后还提出了许多扩展方法[29‒30]。
《3.2 图神经网络》
3.2 图神经网络
除了将分子编码为SMILES字符串外,还可以将分子自然地表示为图。图神经网络(GNN)[31]研究的普及为学习图结构信息(如分子)的表示提供了各种解决方案,在此基础上可以预测反应中的分子转化。Sperduti和Starita [32]是第一个将神经网络应用于有向无环图的人。
研究人员还提出了循环图神经网络(RecGNN),其中邻居信息迭代传播以更新目标节点的表示[33‒34]。受卷积神经网络(CNN)在计算机视觉领域的巨大成功的鼓舞,Henaff等[35]提出了卷积图神经网络(ConvGNN)。以前的ConvGNN有两大类:基于光谱的方法和基于空间的方法。以前的ConvGNN根据定义图卷积的不同方式分为两大类:基于频谱的方法是按照图信号处理来采用滤波器[36‒37],而基于空间的方法是依靠信息传播[38‒39]。后来,图卷积网络(GCN)[40]被开发出来以弥补这些方法之间的差距。最近,GCN由于其效率、灵活性和通用性而继续快速发展。随着注意力机制的广泛应用,一些方法采用注意力机制作为节点聚合机制,如图注意力网络(GATs)[41]和门控注意力网络(GAANs)[42]。在这些工作的基础上,开发了许多替代的GNN方法,包括图自动编码器(GAEs)[43]、图生成网络(GGNs)[44]以及空间-时间图卷积网络(STGCNs)[45]。
《3.3 搜索算法》
3.3 搜索算法
搜索算法检索存储在数据结构中或在搜索空间中计算的信息,多步骤逆向合成预测依靠这些信息来规划合成路线。一般来说,搜索算法可以分为不知情的搜索和知情的搜索。无信息的搜索不利用任何关于状态转换成本的信息;典型的例子包括深度优先搜索和广度优先搜索。相比之下,知情搜索包含启发式函数来评估当前状态和目标状态之间的距离,以指导搜索进度。这保证了在合理的搜索时间内得到一个好的解决方案,尽管该解决方案不一定是最优的。最佳优先搜索是典型的启发式搜索,有一个优先队列的概念[46]。开放列表存储当前可遍历的节点,关闭列表存储已遍历的节点。Beam搜索通过扩展有限集合中最有希望的节点来改进最佳优先搜索[47]。A*搜索结合了统一成本搜索和最佳优先搜索的亮点,确保了最优解[48]。在这种情况下,每个状态的成本是指从起始状态到当前状态的实际成本和从当前状态到目标状态的启发式成本的组合。蒙特卡洛树搜索(MCTS)[49]通过蒙特卡洛模拟提高从当前状态到目标状态的价值估计,包括四个步骤:选择、扩展、模拟和反向传播。AlphaGo [50]是MCTS最有名的使用案例之一,MCTS在围棋的搜索树中搜索可能的动作并跟踪结果。
《3.4 深度强化学习》
3.4 深度强化学习
强化学习(RL)[51]是一种机器学习范式,在这种范式中,代理人与环境互动,并通过试验和错误使累积奖励的概念最大化。RL不需要大规模的注释数据集,并且可以胜任具有良好连续性和创新性的顺序决策问题。最近,RL伴随着深度学习在学习表征方面的快速发展而进步。深度Q网络(DQNs)[52],即使用卷积神经网络来估计Q值,是深度RL领域的一项开创性工作。自其发展以来,研究人员提出了几个基于价值的方法的扩展[53];此外,基于模型的方法[54]和策略梯度方法[55‒56]也被提出,以用预测模型预测采取行动后的状态,并直接优化策略网络。深度RL也适用于更复杂的决策问题,如有目标条件的问题[57]、分层任务分解[58]和多个代理[59]。深度RL已经在游戏[60]、机器人[61]、自动驾驶[62]和分子生成[63]等应用中取得了广泛的成功。它被认为是迈向通用人工智能的重要一步[64]。
《4、 人工智能如何应用于化学逆合成》
4、 人工智能如何应用于化学逆合成
过去十年来,人工智能的空前发展导致最近涌现大量研究,这些研究借助于人工智能技术来解决第2.2节中讨论的挑战,提高逆向合成预测的性能。在图5中,我们对人工智能驱动的逆向合成预测的现状进行了细分,涵盖了单步逆向合成预测和多步逆向合成预测这两个主要部分,以及分子表示和候选反应评估这两个算法设计要素,它们分别为单步和多步逆向合成预测提供了基础条件。这四个要素共同为人工智能驱动的逆向合成预测方法建立了一个设计空间,为新算法的开发和特定应用的定制提供了方向。在这一节中,我们根据提议的格局对现有文献进行细分。
《图5》
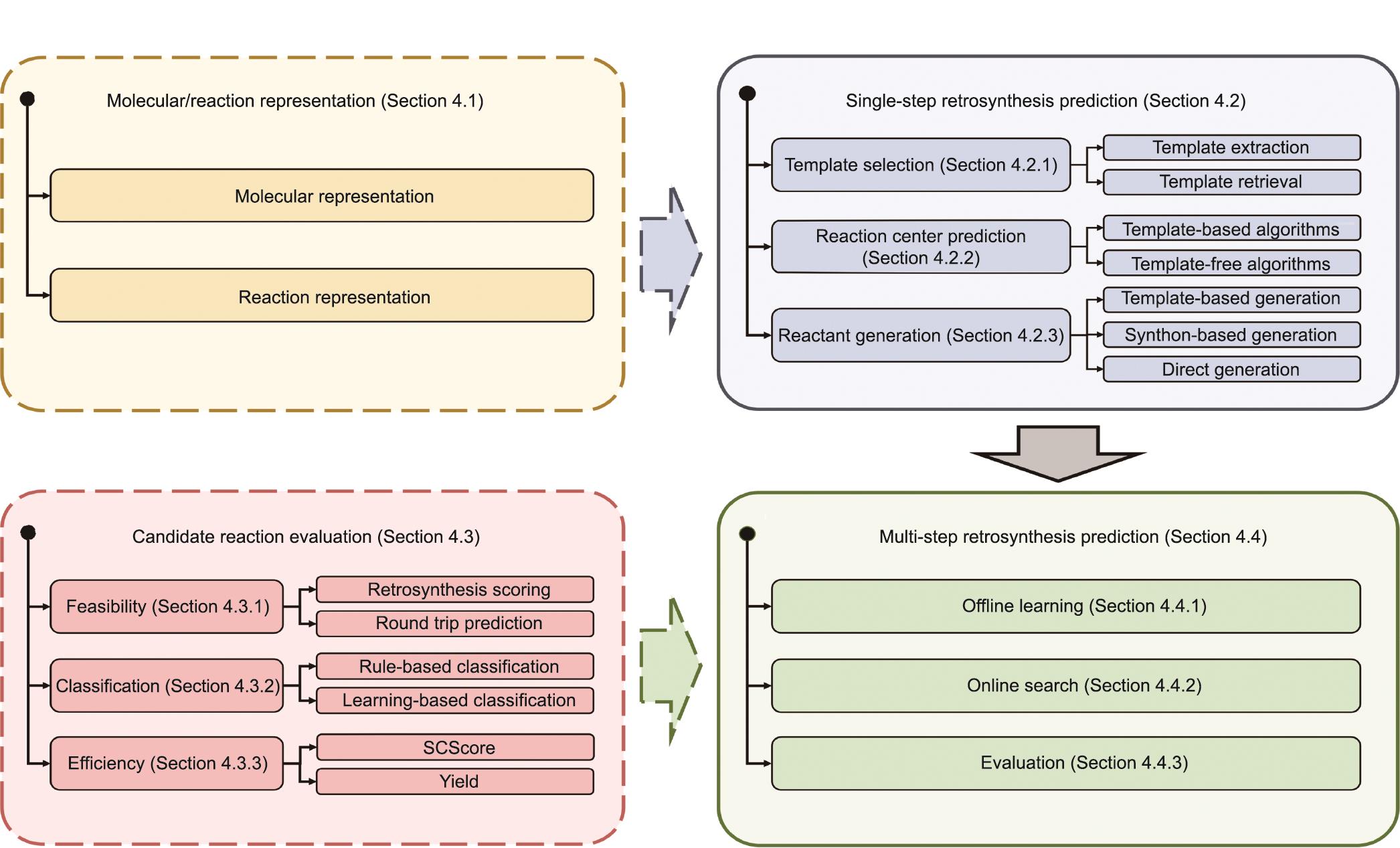
图5 涵盖逆向合成规划算法的两个关键部分的逆向合成预测的概览。右侧是单步逆合成预测和多步逆合成预测,同时还有为这两个部分做准备的两个不可缺少的设计元素:分子/反应表示和候选反应评估(左侧)。
《4.1 分子和化学反应的表示》
4.1 分子和化学反应的表示
《4.1.1. 分子的表示》
4.1.1. 分子的表示
应用于回溯合成问题的主流分子表示方法包括SMILES字符串、指纹和图形,如图6中所总结。SMILES是最广泛采用的分子结构表示方法的化学符号系统[16]。它很容易被化学家所接受,但又有足够的互动性,允许计算机生成独特的线条符号。与自然语言类似,SMILES系统将字符和语法规则结合起来,根据化学原理指定严格的结构[65]。因此,一旦创建了SMILES表示法,研究人员就可以通过序列到序列的机器翻译模型直观地预测用于逆合成的单步前体。
《图6》

图6 用于学习的三种主流分子表示方法的总结:SMILES字符串、摩根指纹向量和图。
分子指纹是另一个有用的化学信息学工具,用于表示分子[66]。它的核心思想是将分子映射成一个长度为
《4.1.2. 化学反应的表示》
4.1.2. 化学反应的表示
化学家把化学反应定义为在试剂的条件下从反应物到产物的转化。因此,表示反应的一个直接方法是将反应物、试剂和产物的字符串连接起来。一个反应的SMILES字符串由反应物、试剂和产物的SMILE字符串以及连接它们以表示反应方向的“>”符号组成[16,65];例如,“反应物>试剂>产物”。同样,分子指纹也可以串联起来作为反应的表示。除了用分子表征以组合方式表示一个反应外,从预训练的模型中提取反应嵌入是另一种有希望的方式[72]。另一种方法是将化学反应放入一个单一的压缩反应图(CGR)中,它是反应物和产物图的叠加[73]。在CGR中,反应中的原子和键的性质变化被突出显示。值得注意的是,反应的CGR表示依赖于反应物和产物之间的原子对原子的映射,CGRTools [74]是一个很好的CGR表示工具。
《4.2 单步逆合成反应预测》
4.2 单步逆合成反应预测
《4.2.1. 模板选择》
4.2.1. 模板选择
如图2所定义的,一个模板编码了产物和反应物之间的连接性变化,而一个模板集包含了一组不同化学规则的模板。给定一个模板和一个匹配的产物,很容易得到反应中的所有反应物。因此,基于模板的方法[10‒11,75‒76]已被规定用于单步逆合成预测。
《4.2.1.1. 模板提取》
4.2.1.1. 模板提取
最早的化学反应规则是由化学家编纂的。专家们制定了哪些反应是允许的[77]。从那时起,手工制定的通用反应规则被用于逆合成预测,并在一些复杂产品上取得了巨大的成功[6,78‒79]。随着反应的爆炸性增长,迫切需要自动提取模板,因此提出了启发式算法,从已知的反应实例中建立通用规则[80‒83]。其关键思想是提取反应中心并包括不同数量的相邻原子。邻近原子的数量由与反应中心的固定距离或哪些原子相关的启发式方法决定。不同的数量决定了模板的不同泛化水平。为了处理立体化学模板,Coley等[84]提出了RDChiral——一个RDCit [85]包装器,用于逆合成模板提取。
由专家定义的规则是精确的,但数量太少,无法覆盖足够多的反应类型。虽然通过自动模板提取创建模板的效率很高,但这种模板在通用性和正确性之间存在折中。此外,模板的数量是巨大的[84],并继续增加[86],因为新的反应被不断发现。
《4.2.1.2. 模板检索》
4.2.1.2. 模板检索
在提取模板之后,还有一个研究问题:在给定目标产品的情况下,如何从模板库中检索出模板。RetroSim [11]的方法模仿了化学家在逆向合成预测过程中可能的决策过程;也就是说,它模仿了类似分子的合成过程。编码结构与目标分子相似的分子的合成路线的模板被优先考虑,并且只检索其产品与目标分子相似度
NeuralSym [76,91]不依赖于明确的分子相似性,而是将模板检索问题作为一个多类分类问题,以便自动学习目标分子的模式,从而决定使用哪个模板。如图7所示,一个全连接的神经网络被用来进行多类分类。神经网络的输入是用摩根指纹表示的目标分子[69],输出节点对应于所有的模板。当得到一个新的目标分子时,训练有素的神经网络会检索出其softmax概率最高的前
《图7》
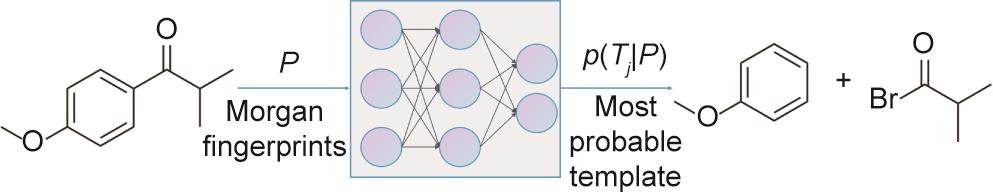
图7 神经符号(NeuralSym)模型的说明。
尽管NeuralSym被训练成在基准数据集中学习给定分子的单一模板,但基准之外的许多潜在的有用的反应被排除在外[75]。为了解决这个问题,Fortunato等[75]建议用更多关于模板适用性的合成数据来增强基准,并预训练一个模板相关性网络,学习所有可能导致反应物的模板。预训练的模板相关性网络,后来在真实反应中进行了微调,在模板适用性方面实现了更高的召回率,并且比以前的方法(如NeuralSym)在生成的反应物方面有更多的多样性。另一个名为MHNreact [92]的替代方法通过编码模板和产物作为输入来解决这个问题,与之前的方法不同的是,当给定目标分子作为输入时,它预测的是一套固定模板。然后,MHNreact采用现代霍普菲尔德网络(MHNs)来学习模板和产品之间的关联。
NeuralSym的一个明显的局限性是,模型的大小随着模板数量的增加而增长。为了克服这一限制,Dai等[10]提出了条件图逻辑网络(GLN)。GLN通过学习模板和反应物的编码直接对它们的条件联合概率进行建模和最大化,即
《4.2.2. 反应中心预测》
4.2.2. 反应中心预测
《4.2.2.1. 基于模板的方法》
4.2.2.1. 基于模板的方法
基于模板的算法只需应用检索到的模板并生成反应物。检索到的模板提供了几个候选反应中心。RetroSim [11]和NeuralSym [76]根据模板的等级来确定反应中心,而GLNs [10]根据反应物的等级来预测反应中心,如上所述。
《4.2.2.2. 无模板的方法》
4.2.2.2. 无模板的方法
尽管有可解释性和实质性的进展,但需要子图同构的基于模板的算法仍然存在可扩展性差和对新反应的概括性差的问题。最近,围绕着绕过模板的主题,出现了大量的文献,这些无模板算法使用机器学习模型来直接预测反应中心。
反应中心的识别问题归结为链路预测,即预测分子图
《4.2.3. 反应物生成》
4.2.3. 反应物生成
《4.2.3.1. 基于模板的生成》
4.2.3.1. 基于模板的生成
反应物是通过子图匹配得出的,给定一个目标产品和一个模板[84]。RetroSim [11]和NeuralSym [76]根据模板的得分进行排序,并按照前k个模板的相同顺序生成前k个反应物。GLNs [10]对选定的模板应用于目标分子后的所有反应物进行排序。反应物的分数
《4.2.3.2. 基于合成子的生成》
4.2.3.2. 基于合成子的生成
在G2Gs [17]和RetroExpert [8]中,一组中间合成子
《4.2.3.3. 直接生成》
4.2.3.3. 直接生成
分子编辑图注意网络(MEGAN)[97]直接从目标分子的图表示中生成反应物
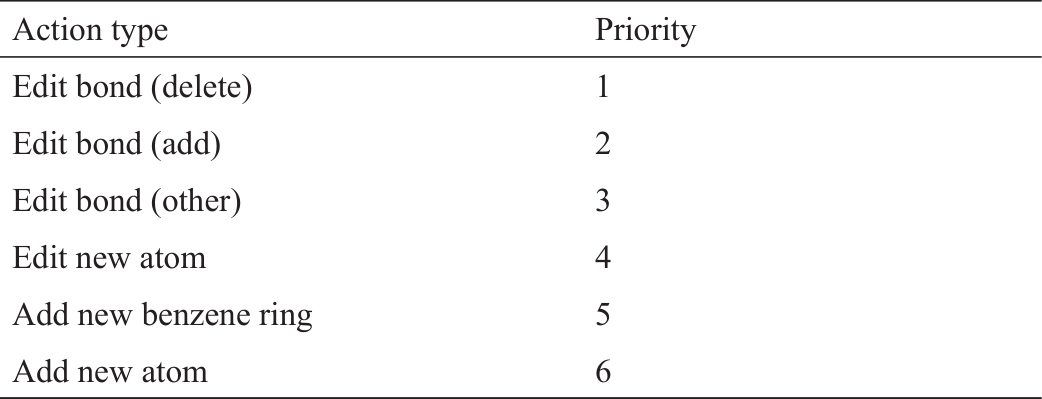
《表1》
表1 用于训练MEGAN的行动优先顺序[]
除了GNN,其他研究者将单步逆合成预测表述为一个序列到序列的机器翻译问题,其中输入是目标分子的SMILES字符串[16],输出是所有反应物的SMILES序列。如图8所示,Liu等[7]采用biLSTM [99]作为神经序列到序列型。为了利用神经机器翻译的最新进展,提高逆合成预测的准确性,Karpov等[100]用Transformer模型[28]取代了biLSTM。
《图8》
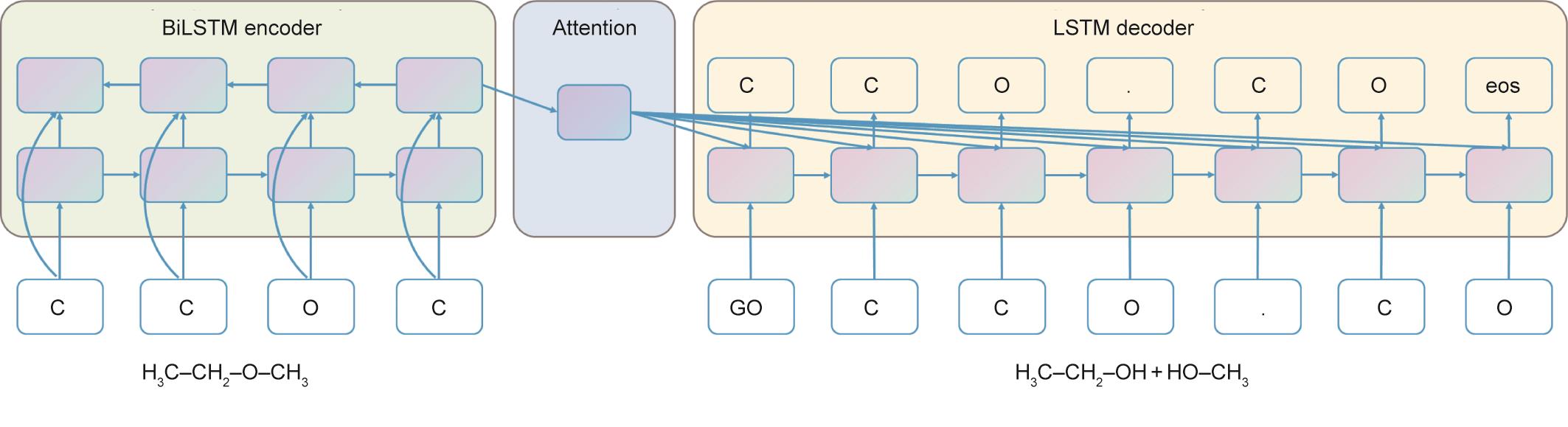
图8 用于逆向合成预测的biLSTM模型的说明。
序列到序列的翻译算法的主要弱点是它们的性能不令人满意,这有三个原因:第一,将分子表示为字符串忽略了丰富的化学结构和原子之间的相互作用;第二,生成的SMILES字符串的化学有效性得不到保证;第三,端到端的训练策略未能包括丰富反应中的任何化学知识。除了性能差之外,序列对序列模型在预测方面缺乏可解释性。另一方面,直接生成享有以下独有的好处:①虽然基于模板的算法生成试剂(包括溶剂、催化剂和其他物质)以导致反应的发生是具有挑战性的,但对于直接生成来说是有可能的;②与基于合成体的生成相比,直接生成倾向于产生更多样化的反应,这对于多步骤的逆向合成是至关重要的。今后,有希望通过数据的增加和丰富来提高序列对序列方法的性能。最近,基于模板的双重建议(DualTB)[101]通过制定一个新的基于能量的框架,在提供无模板方法和基于模板方法的统一观点方面取得了进展。DualTB通过定义不同的能量函数,轻松容纳了这两类方法;在推理阶段,通过模板匹配获得的或由模型生成的反应物候选物根据预测的能量得分进行排序。
《4.3 候选反应评估》
4.3 候选反应评估
《4.3.1. 可行性评估》
4.3.1. 可行性评估
《4.3.1.1. 逆合成评分》
4.3.1.1. 逆合成评分
逆向合成模型本身能够用不同的分数对预测的反应进行排名。例如,RetroSim [11]使用分子相似性对候选化学反应进行排名;总体相似性得分是产物相似性(
《4.3.1.2. 往返预测》
4.3.1.2. 往返预测
对预测的逆向合成可行性的严格评价是调查所产生的前体(即反应物)是否能按预期产生产品,这就需要一个正向化学反应预测模型。Schwaller等[15]创造了“往返评价”一词来描述这种评价,并引入了两个往返评价指标:往返准确性和覆盖率。往返准确性指的是有效反应物与后向化学反应预测模型所预测的所有反应物的比率,其中“有效”意味着反应物可以产生目标产物,正如前向化学反应预测模型所预测的那样。覆盖率评价的是逆向合成模型产生至少一组有效反应物的目标产品的数量。覆盖率评价指标鼓励逆向合成模型为各种各样的目标分子产生有效的反应物,从而弥补了往返准确性的缺陷,后者可能过度奖励那些只为少数反应产生许多有效前体的模型。
《4.3.2. 多样性》
4.3.2. 多样性
在化学反应空间中,有许多生成分子的方式,不同的方式可能对应于不同的反应类型。这里需要指出的是,具有相同反应机理的有机反应属于同一类型。一个好的逆向合成模型应该为目标分子产生尽可能多的合成路线,其中多样性可以通过属于
(2)
式中,
《4.3.2.1. 基于规则的反应分类》
4.3.2.1. 基于规则的反应分类
传统上,反应都是根据产物的类别、官能团、使用的试剂甚至是反应的发明者进行手工分类。然而,使用这种分类方法对反应的覆盖面是远远不够的。后来提出了基于模型的方法,该方法依赖于反应中心的预定义定义,并考虑反应过程中变化的键[102]。作为克服基于模型的方法忽视反应中心以外的功能和子类的限制的一种手段,数据驱动的方法[103]通过扩展反应中心以包括相邻的键来识别反应中心的多层次描述。考虑到原子类型、价态、成键氢原子总数、π电子数、芳香度、形式电荷和反应中心信息,自动为反应中心的每一级描述计算哈希代码——也称为反应类型。另一种数据驱动的方法[104]将共享反应分类(RC)编号的反应归为一类,其中RC编号表征了反应中心原子类型的转化模式。凝聚反应图(CRG)方法[73]直接将一个反应的所有反应物和产物合并为一个假想的过渡态或伪分子,这样就可以用伪分子的描述符来评价反应之间的相似性。然而,基于数据驱动的方法很可能无法检测到具有相似的基本机制但扩展反应中心的拓扑结构不同的反应。
使用已知反应机制或反应的大型规则库,NameRXN [105]能够对大约1000个名称反应进行分类。虽然它很详细和准确,但NameRXN通常只能识别大约50%的反应的类别。因此,有必要使用基于学习的分类算法来归纳其余未分类的反应。
《4.3.2.2. 基于学习的分类》
4.3.2.2. 基于学习的分类
越来越多的大型反应数据集促进了机器学习算法的发展,对化学反应进行分类,其中分类器是由NameRXN直接用一组分类的反应进行训练。Schneider等[106]研究了一系列作为特征的反应差异指纹和五种机器学习模型,包括随机森林、天真贝叶斯、K-均值、逻辑回归和K-近邻,用于对名称反应本体(RXNO)中的50种反应类型进行分类[103]。后来在文献[107]中提出了一个分层分类模型。其中共形预测(CP)框架被用来预测具有置信度的层次结构中的336种反应类型,并有信心措施。最近,Schwaller等[72]使用一个名为RXNFP的基于Transformer的模型,实现了98.2%的最先进的分类准确率,只使用化学反应的SMILES表示作为输入,并且不需要对反应中心进行自动标注。
《4.3.3. 有效性》
4.3.3. 有效性
评价一个反应是否应该被优先考虑的另一个理想品质是效率,它是用反应物的合成复杂性得分和目标产物的产量来衡量的。更容易合成的反应物和更高的目标产品产量的反应是首选。
《4.3.3.1. SCScore》
4.3.3.1. SCScore
Coley等[108]提出了一种计算反应物的合成复杂性的方法。SCScore的关键思想有两个方面。首先,如果一个分子看起来需要很多反应步骤才能用传统方法合成,那么它就被认为很难制造,并被赋予较高的SCS分数;其次,一个分子作为反应物出现的频率越高,其合成复杂性就越低。应该注意的是,反应物的SCS分数总是小于或等于产品的SCS分数。
《4.3.3.2. 产率》
4.3.3.2. 产率
在一个化学反应中,有可能有些反应物不会反应生成产物。化学反应的产率描述了成功转化为产物的反应物相对于理论最大值的百分比:
(3)
式中,y表示产率;
最近,人们对使用机器学习方法预测产量的兴趣越来越大。Skoraczyn´ski等[110]基于通过分子指纹和化学语言描述符(CLD)来表示反应[109],比较了各种机器学习方法(如决策树和随机森林)来预测反应产量。受最近基于Transformer的模型在反应类型分类中取得的成功的启发[72],Schwaller等[111]对RXNFP模型[72]进行了微调以预测产量。遗憾的是,由于产量预测问题带来了定义不明确和噪声注释的挑战,其性能并不十分令人满意。
《4.4 多步逆合成路线规划》
4.4 多步逆合成路线规划
当致力于多步逆合成计划时,机器需要从合法的候选集合中选择一个反应,在每一步给定一个目标分子,观察反馈,然后在一个新的目标分子上进行下一个选择动作。这个过程与一般战略决策游戏中的顺序决策有着惊人的相似性;换句话说,多步骤逆向合成计划的问题可以被设定为一个单人游戏[112]。因此,多步骤逆向合成规划中必须解决的两个核心研究问题与解决博弈的问题相同,即如何有效地搜索成功的路线以及如何估计每条路线的价值。此外,与国际象棋等游戏不同的是,获胜状态是二进制的,而逆向合成规划需要根据合成的成本来区分获胜状态。剩下的第三个艰巨的研究挑战是评估合成成本,它涉及多个数量,如合成步骤的数量、产量和商业可用反应物的总成本。
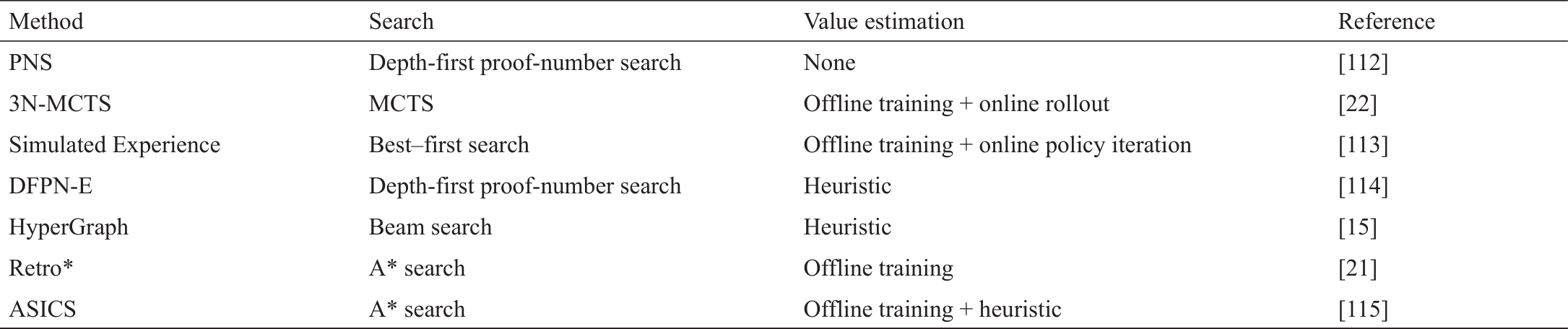
在下一节中,我们将回顾表2中总结的最新方法[15,21‒22,112‒115],并仔细研究它们如何以离线方式学习先验值估计神经网络(离线学习),如何进行在线搜索,其中估计值可以选择细化(在线搜索),以及如何评估一个逆合成路线(评估)。
《表2》
表2 关于两个中心研究问题的多步骤逆向合成规划算法的现有方法的总结
《4.4.1. 离线学习》
4.4.1. 离线学习
由于很难启发式地准确确定一个分子的逆向合成价值(即成本),研究人员有动力从历史上模拟的逆向合成规划经验中学习该价值。以这种离线方式用神经网络参数化的学习策略作为搜索算法的可靠先验概率。当然,关键是要构建一个逆向合成规划路线的训练数据集,其中每个分子都被注释为其合成路线的定义成本。现有的工作依赖于化学家设计的路线或用单步反应数据集手工制作的路线;因此,已建立的逆向合成路线数据集由于只基于成功的路线而存在规模有限、噪声和不平衡的问题。值得注意的是,Segler和Waller [22]使用文献中的反应作为基础事实来预训练一个神经网络,该网络在给定一个特定的分子时确定生成逆合成反应的反应模板的等级。策略网络后来被纳入置信度上限(UCB)公式中,以平衡MCTS中的探索和利用。
相反,Chen等[21]没有直接使用文献中的反应,而是手工制作了一个逆合成路线的数据集,用于预训练策略网络。他们对出现在单步反应数据集中的每个分子进行分析,检查该分子是否能被数据集中的现有反应所合成,并连接合成该分子的最短路线。所有的路线都被收集起来,以便对估计合成分子价值的策略网络进行预训练,这被用作A*搜索中的启发式方法。Schwaller等[15]使用预训练的反应复杂性指标,如SCScore [108],作为启发式方法,形成类似贝叶斯的概率作为探索的先验知识。
《4.4.2. 在线学习》
4.4.2. 在线学习
在线搜索是逆向合成规划中最重要的组成部分。人工智能中有许多搜索算法,从贪婪的深度优先搜索等非知情算法到证明数字搜索(PNS)和A*等知情算法。现有的关于多步骤逆向合成预测的研究偏向于知情算法,因为使用启发式函数有可能产生好的解决方案,所以其更有效率和准确性。然而,启发式函数的定义并不复杂,因为在整个合成路线完成之前,每个反应的价值都是不确定的,而好的合成路线的定义是模糊的和主观的。例如,带有三个神经网络的MCTS(3N-MCTS)[22]使用传统的UCB公式作为启发式,以达到探索和开发之间的平衡。除了一个预训练的策略网络(它只决定了开发)外,3N-MCTS还通过其快速展开的策略网络纳入估计值,以完善UCB中的启发式功能。图9展示了通过3N-MCTS进行逆向合成规划的四个阶段,图10提供了一个返回搜索树的示例。HyperGraph搜索[15]将反应置信度得分与3N-MCTS和SCScore中使用的相同的单步逆合成模型相结合,形成类似贝叶斯的概率,作为波束搜索的启发式。作为一种替代方法,PNS [116]是一种有效的搜索算法,用于解决游戏,特别是解决困难的残局位置。Heifets和Jurisica [112]通过将逆向合成规划建模为一个双人游戏,直接将深度优先证明数搜索(DFPN)应用于基于AND-OR树的多步逆向合成预测。Kishimoto等[114]修改了启发式方法,以避免启发式在片面的搜索空间中退化。Chen等[21]将PNS和A*启发式搜索结合起来进行逆向合成规划,其中预训练的策略网络被用来获得每个未解决分子的逆向合成成本的预先估计。同样,Jeong等[115]对预定的反应知识图和基于机器学习的反应预测进行了类似A*的混合搜索,其中启发式被设计为包括分子相似性、合成可及性得分和可能性得分。
《图9》
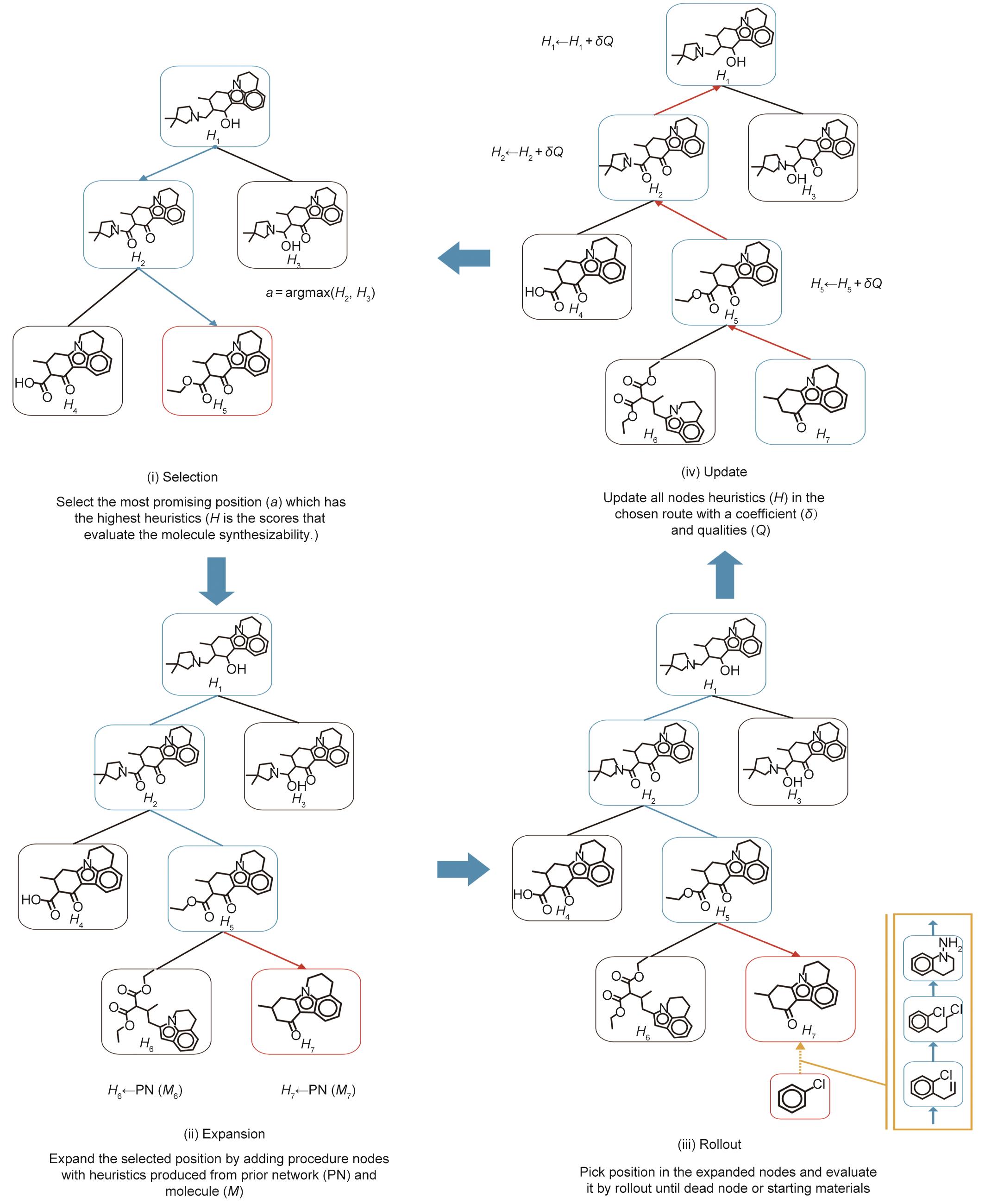
图9 逆向合成规划的MCTS的四个阶段的说明。
《图10》
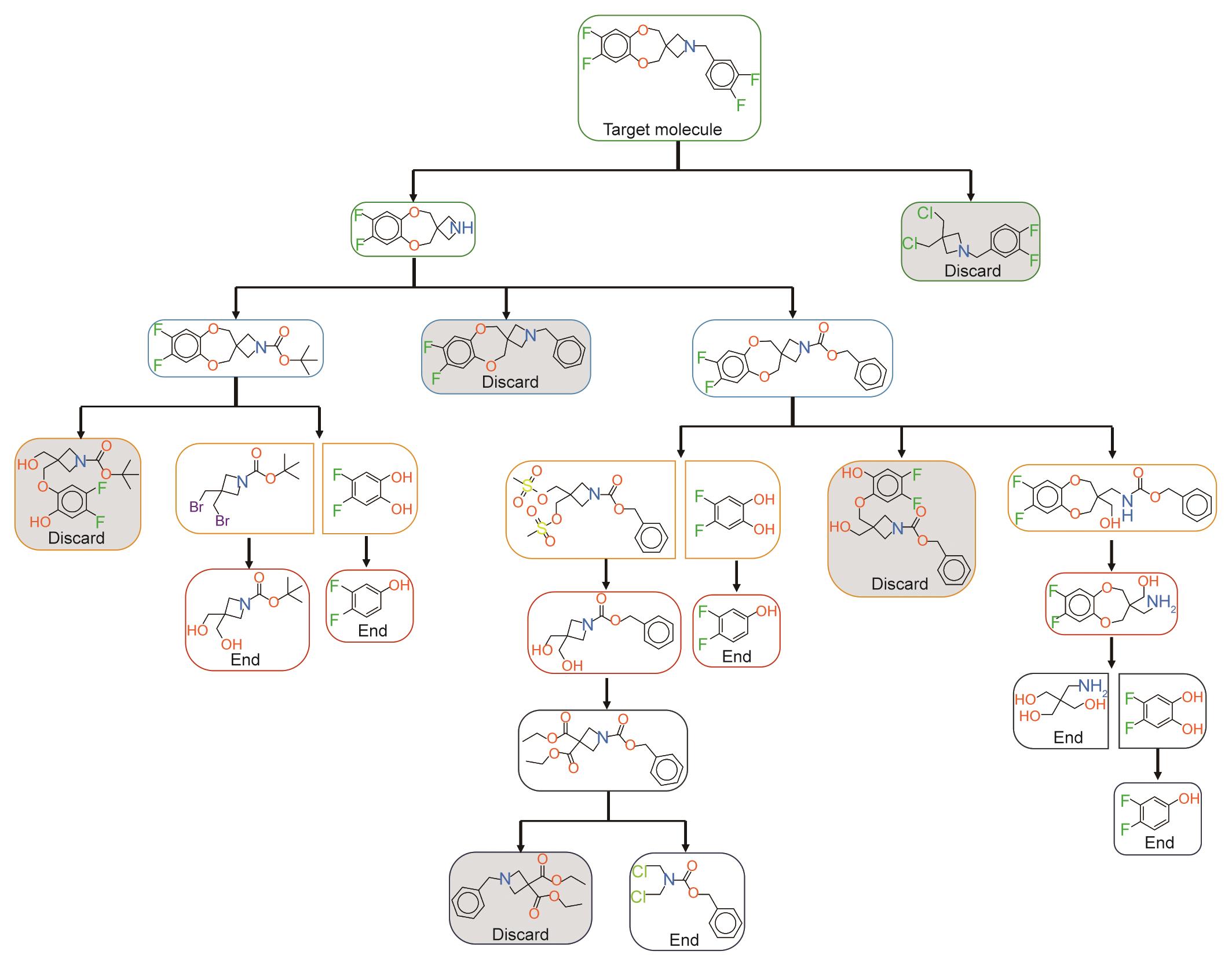
图10 一个示例性的逆向合成搜索树。搜索树从目标分子开始;树上的每个节点要么被丢弃,要么被拾起进行扩展;当一个叶子节点没有子节点可以扩展或到达起始材料时,它就结束了。
《4.4.3. 评估》
4.4.3. 评估
对一个逆向合成规划模型的评价往往可以用不同的、多维的方式进行。在此,我们把对搜索算法的评价分为两个主要指标:效率和质量。在效率方面,Chen等[21]将单步逆向合成模型的调用次数作为合成所需时间的代用指标。两种普遍采用的方法被用作质量的衡量标准:①评估任意一组分子在固定的单步推理调用次数内最终完成逆合成途径(即到达起始材料)的成功率;②评估一条途径的成本和长度。评估成本的一种方法是计算沿途所有单步反应的负对数可能性(NLL)的总和。路线的长度只是相当于给定路线中反应的总数。超图搜索[15]将化学选择性引入路线评估。遗憾的是,目前还没有一个自动工具来量化一个反应的化学选择性的准确性。
《5、 已有工作综述》
5、 已有工作综述
《5.1 单步逆合成反应预测》
5.1 单步逆合成反应预测
《5.1.1. 数据集》
5.1.1. 数据集
源自美国专利商标局(USPTO)授予的专利[117],USPTO-50K [118]和USPTO-full [119]是两个广泛采用的单步逆合成预测的基准数据集。USPTO包含50K反应,涵盖10种反应类型,USPTO-full由USPTO 1976‒2016年的950K纯化反应组成,不受限于特定的反应类型。正如文献[7‒8]所建议的,USPTO-50K和USPTO-full都有8∶1∶1的随机训练/验证/测试分割。
《5.1.2. 模型和训练细节》
5.1.2. 模型和训练细节
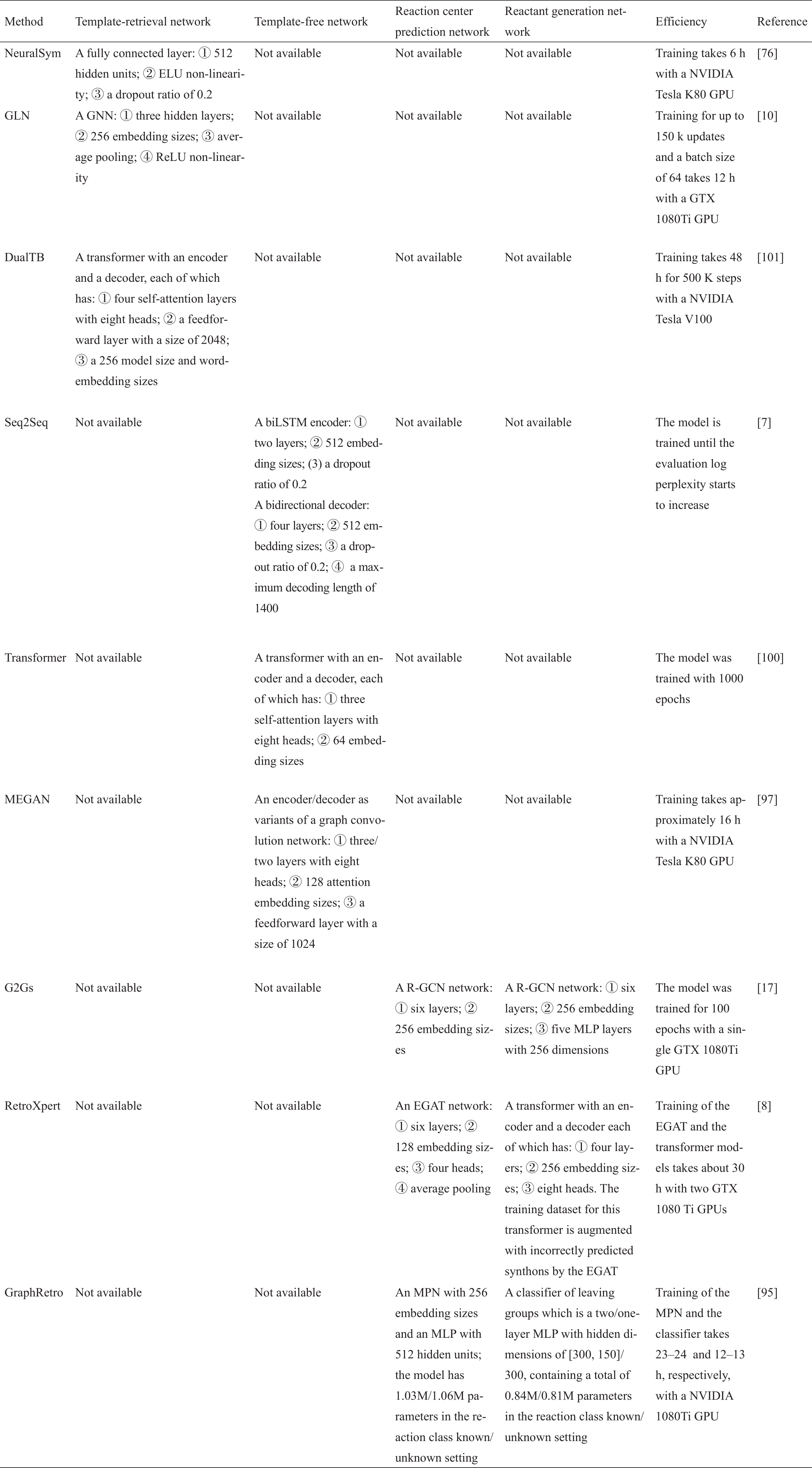
表3 [7‒8,10,17,76,95,97,100‒101]列出了每个单步逆合成预测算法的具体架构和训练效率。前三个基于模板的方法构建了模板-检索网络,接下来的一批方法(Seq2Seq、transformer和MEGAN)构建了序列或图的翻译模型。最后三种算法是基于半模板的,因为它们首先建立一个反应中心预测网络来预测反应中心,然后构建一个反应物生成网络来将合成物转换为反应物。
《表3》
表3 现有单步逆向合成预测算法中的架构和训练细节总结
《5.1.3. 评估指标》
5.1.3. 评估指标
top-k精确匹配的准确性是一个常用的评估指标,用于评估地面真相的反应物是否落入top-k预测的反应物列表中。准确率的计算方法是将预测的反应物的标准SMILES字符串与ground-truth进行匹配,通常使用RDKit [85]包进行canonicalization。值得注意的是,RDKit是一个广泛流行的开源化学信息学工具箱。
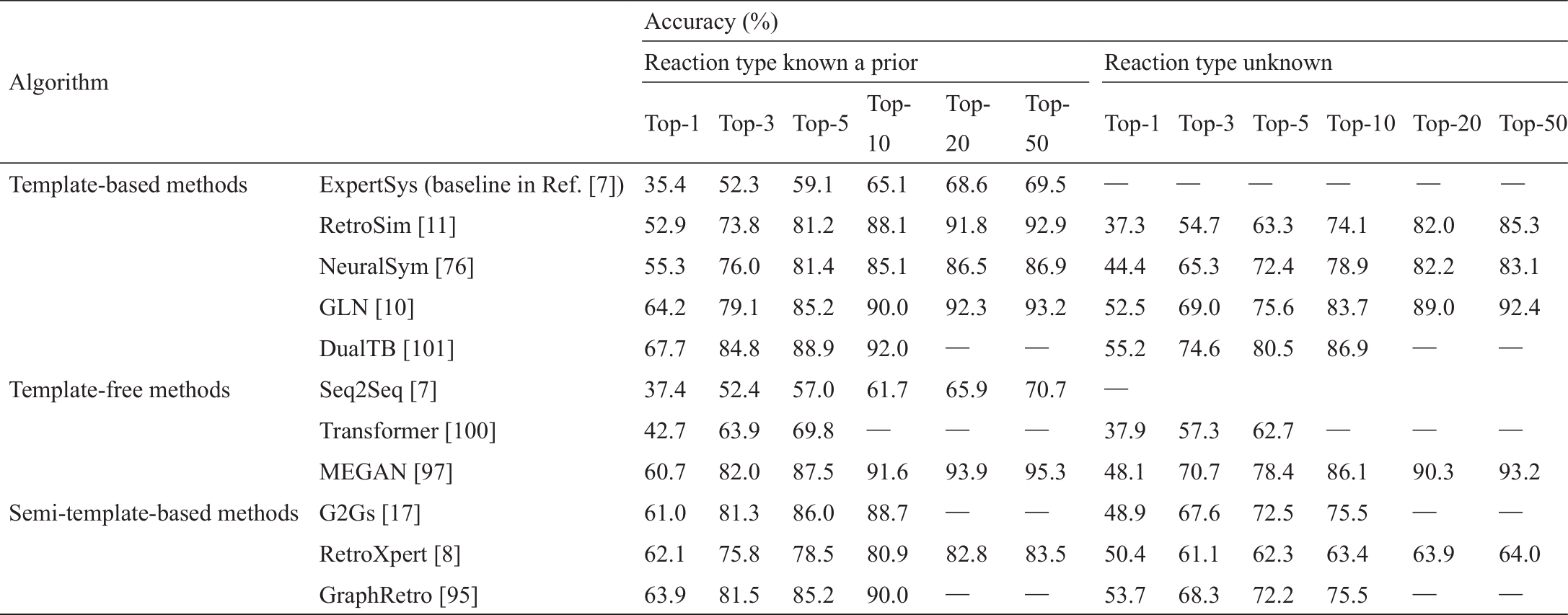
表4 [7‒8,10‒11,17,76,95,97,100‒101]总结了基于模板和无模板算法在USPTO-50K上的结果,其中基于半模板的方法是指两阶段无模板算法,其先预测反应中心,然后生成反应物。我们将主要结论列举如下:首先,基于规则的算法,如ExpertSys,表现得很差;其次,基于模板的方法——所有这些方法都使用参考文献[10]中的同一套模板,是相当有竞争力的——超过了序列到序列的无模板方法;再次,在识别反应中心时,两阶段的无模板算法往往是无模板算法中表现最好的,特别是对top-1的预测;最后,如果不费吹灰之力就能自动提取模板,那么GLN在前5/10/20/50名的预测中更受欢迎。鉴于实践中的目标分子与模板中的分子有很大的不同,基于半模板的方法具有卓越的概括能力和竞争性的预测能力,是首选。在表5 [10‒11,76,97]中,我们展示了四种选定的算法在USPTO全数据集上的可扩展性。可以看出,GLN和DualTB的表现超过了基于相似性的模板检索方法RetroSim和基于分类的模板检索方法NeuralSym。
《表4》
表4 USPTO-50K上单步逆合成预测的现有算法比较
《表5》
表5 USPTO-full上单步逆合成预测的现有算法比较

《5.2 多步逆合成路线规划》
5.2 多步逆合成路线规划
正如第4.4节所讨论的那样,评价一个逆向合成规划路线的质量是特别具有挑战性的。首先,评价指标是模糊的。化学家们认为一条好的合成路线可能是因为采用了短的途径或廉价的起始材料,以及具备容易满足的反应条件等。其次,没有大规模的基准数据集,包括良好的合成路线,可以用来定量比较不同的多步骤逆合成预测算法的性能。下面是一些数据集和评价指标,作为折中的方案。
《5.2.1. 数据集》
5.2.1. 数据集
《5.2.1.1. 有专家标注的小规模基准测试》
5.2.1.1. 有专家标注的小规模基准测试
由于难以收集已发表的论文中证明有效的合成路线,Heifets和Jurisica [112]构建了一个小型的基准,包括20个出现在马萨诸塞州理工学院化学课程考试中的合成问题。答案是由指导教师提供的。这个基准的主要缺点是它的规模极小。
《5.2.1.2. 带有自动标注的大规模基准测试》
5.2.1.2. 带有自动标注的大规模基准测试
Chen等[21]在USPTO数据集[117]的基础上构建了一个大规模的逆合成路线基准数据集,该数据集由一步反应组成。一个分子的合成路线是通过迭代应用USPTO中的一步反应而自动构建的;如果发现有多条路线,则选择最短的一条。总共有299 202条训练路线、65 274条验证路线和189条测试路线。然而,这个数据集的缺点是USPTO所涵盖的化学反应有限,而且按长度评价路线质量。
《5.2.1.3. 无标注的大规模基准测试》
5.2.1.3. 无标注的大规模基准测试
在参考文献[22,114]中,作者分别直接选择了2015年或之后首次报道的497个不同的分子作为已知的起始分子和897个不在起始分子集合中的目标分子。
《5.2.2. 评估指标》
5.2.2. 评估指标
《5.2.2.1. 化学家的双盲AB测试》
5.2.2.1. 化学家的双盲AB测试
对于一个提供了专家指定的合成路线的目标分子,请化学家通过双盲AB测试直接评估算法预测的路线的好坏[22]。他们需要根据个人参考和合成的可行性在两条路线中选择一条。
《5.2.2.2. 有效性指标》
5.2.2.2. 有效性指标
不同的逆向合成规划算法相互比较,并与通过人工或自动注释开发的路线(如果有的话)进行比较。文献中的有效性指标包括被解决的分子的百分比或数量、路线的长度,以及总结路线上所有反应的NLL的总成本。通常情况下,逆向合成规划算法将单步逆向合成模型预测的每个反应的置信度得分作为对数可能性。另一方面,Mo等[120]提出了一种创新的数据驱动的路线评估方法,该方法提出了一个动态树状结构的长短时记忆(LSTM)模型,将具有不同结构的路径编码到一个嵌入表示。嵌入包括路径层面的信息,这些信息可用于策略路径评估和路径相似性检查。
《5.2.2.3. 效率指标》
5.2.2.3. 效率指标
除了有效性之外,搜索算法的效率也是人们关注的重点。现有的工作评估解决一个分子的时间成本和展开的节点数量。
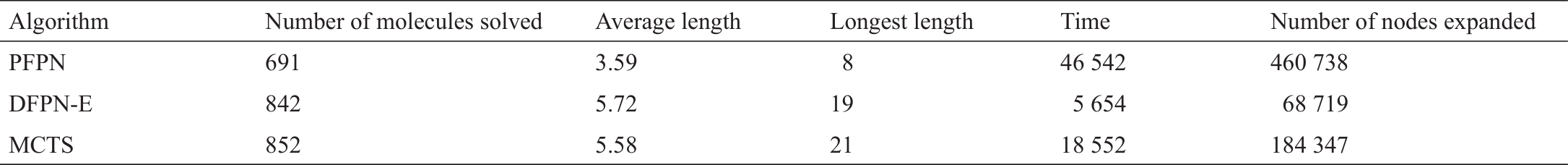
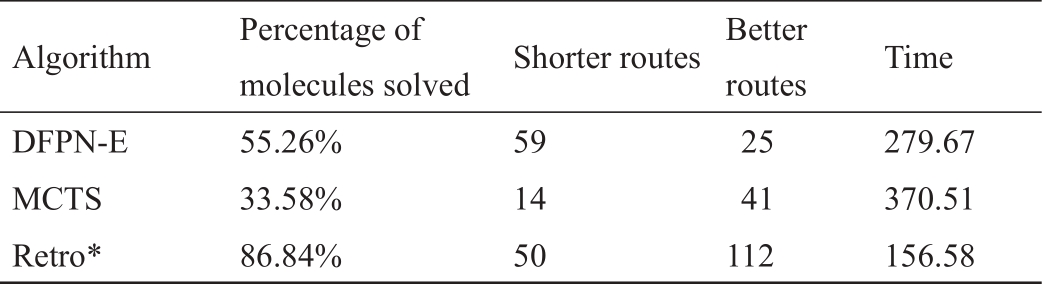
尽管没有对我们所介绍的所有多步骤逆合成预测算法进行一致和全面的性能比较,但我们已经介绍了在其他地方发表的选定算法的性能比较。仔细观察表6 [114]和表7 [21])可以发现,基于AND-OR树的搜索算法,包括Retro*和带有启发式边缘初始化的PNS变体深度优先证明数搜索(DFPN-E),在路线长度和搜索运行时间方面一直优于MCTS。尽管Retro*在表7 [21]中表现突出,我们鼓励在其他数据集上进行更多的比较。用于训练Retro*的价值估计神经网络的训练路线和测试路线是由同一个数据集构建的。在这种情况下,Retro*的表现优于其竞争对手也就不奇怪了。我们还建议对策略迭代算法[113]进行更多的实证研究,与Retro*相比,该算法提高了价值估计的能力。更高级的尝试可能涉及采取两种算法的最佳部分,即基于文献[113],并按照文献[21]用A*搜索。
《表6》
表6 897个无标注目标分子的性能比较[]
《表7》
表7 189个自动标注测试分子的性能比较[]
《5.3 成熟的逆合成规划框架》
5.3 成熟的逆合成规划框架
自1959年Corey和Wipke [4]推出第一个软件以来,计算机辅助逆向合成一直是一个非常活跃和蓬勃发展的研究领域。先锋LHASA [121‒122]和SECS [123]不断激励着逆向合成的发展。遗憾的是,这两个软件包和它们基于规则的后续产品[6]存在两个重大挑战:①手工编码的规则规模和多样性有限,因为扩展规则集是非常费力的;②缺乏对路径进行排名或打分的整体指标。这就促使人们自动生成反应规则,ARChem Route Designer [83]的框架就是一个代表。但是,ARChem未能将立体化学和区域化学考虑在内。
最近,深度学习和人工智能技术提供了巨大的机会,培养了几个优秀的、受欢迎的成熟的逆向合成计划框架,如表8 [13‒15,124‒126]中所总结的。Synthia [13,124],以前被称为Chematica,诉诸于化学评分功能和反应评分功能来决定应用哪一个手编规则(在有经验的化学家提供的100 000个手编规则中)和选择哪一条途径。应该指出的是,这两个函数可以由用户编辑。Synthia通过进行类似于多光束的搜索来探索合成空间。在参考文献[13]中,Synthia被认为是在合成空间中进行搜索的。Synthia被证明能够为多种药物类分子提出实验上成功的途径。尽管在Synthia中使用了手工编码的规则或模板,但值得注意的是,模板的适用性被进一步微调,以通过机器学习模型(即分类器)评估位点或区域选择性。
《表8》
表8 最近成熟的逆向合成规划框架的总结
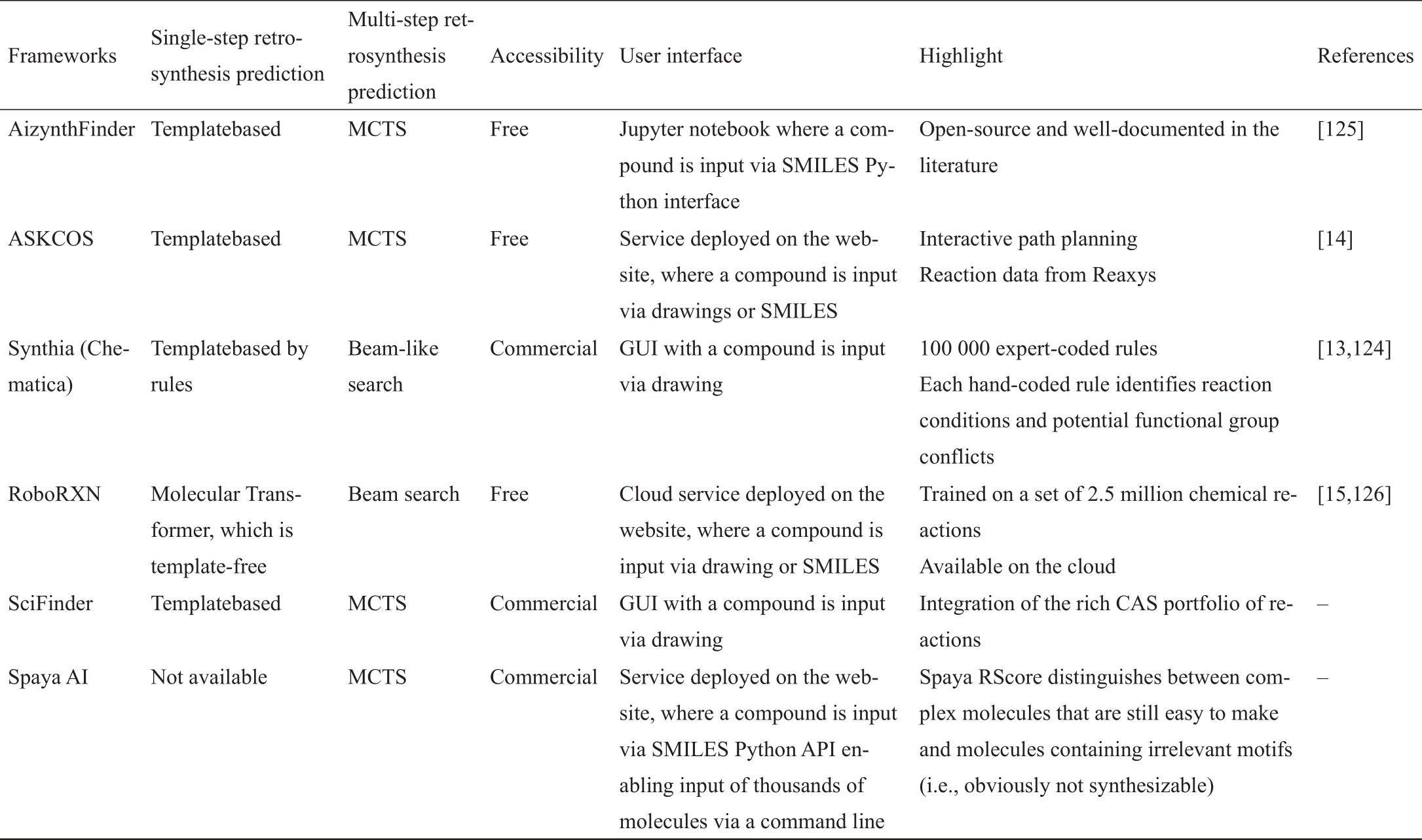
ASKCOS [14]不是商业化的,而是开源的,与以前的框架相比,它更多地是由人工智能驱动的。ASKCOS从Reaxys数据库中自动提取163 273个模板,并训练一个前馈神经网络来预测哪个模板适用于目标分子。对于多步逆合成计划,ASKCOS采用了根平行化的MCTS。为了保证返回的路径的合理性和质量,ASKCOS采用文献[22]中提出的范围内过滤器去除低质量的路径,并去除无法使用的路径,以及无法通过正向预测模型生成目标产品的路径。提出的15个药物分子的合成途径已经通过一个完全自动的机器人系统完成,试剂和反应物很容易获得。
另一个开源软件AiZynthFinder [125],在算法方面与ASKCOS相似,但旨在提供一个灵活的开源基准,支持在其上不断开发。RoboRXN [15,126]正在推进单步逆合成预测,从基于模板的方法到完全无模板的分子转化器。更令人印象深刻的是,RoboRXN首先引入了四个新的指标,包括覆盖率、类多样性、往返准确性和JSD,以彻底评估分子转化器;在这些指标的帮助下,构建了潜在反应物的超图,并在超图上进行波束搜索,得出路径的结论。IBM的研究人员已经证明,RoboRXN可以在短短1 h内按照自己计划的途径合成3-溴苄胺。其他框架,如SciFindern和Spaya AI,是商业性的,部分未公开。
这些成熟框架的可及性主要由反应数据库或手工编码的反应规则(模板)的可用性和起始材料数据库(如eMolecules)的可用性所决定。商业框架依赖于商业数据库,而开源框架则借助于公开可用的数据库。表8显示,需要最低限度专业知识的界面(如输入SMILES和绘制分子)是相当友好的,以方便化学家采用这些逆合成框架。我们希望目前最先进的单步逆合成预测算法(如DualTB [101])和最先进的多步计划方法(如Retro* [21])能够取代这些现有框架中的对应算法,特别是开源的ASKCOS和AiZynthFinder,从而使计划的途径在学术界或商业用途中取得更多令人兴奋的突破。
《6、 结论与展望》
6、 结论与展望
在过去的三年里,机器辅助的逆向合成规划得到了快速发展。纯粹的面向数据的人工智能方法[22]在有效性和效率方面都取得了重大进展,超过了基于提取规则和手工设计启发式方法的传统机器。不断发展的混合专家-人工智能系统称为Synthia [13,124],在规划复杂目标的合成方面补充了面向数据的方法。开源框架ASKCOS [14]甚至推动了机器人执行计划的化学合成路线,进一步节省了专家化学家的时间和精力。这项工作旨在对算法设计空间中现有的逆向合成方法进行全面回顾,以便:①让计算机科学家和计算化学家了解在药物化学目标的逆向合成中所探索的和剩余的研究问题。鼓励将最先进的机器学习方法整合或发展为成熟的框架,提供对化学家友好的界面,并且仍然是化学家的首要选择;③激励更多的研究,以改善整个设计空间的任何研究方面,如反应表示或反应中心预测。在机器被广泛接受为化学家的有用助手之前,仍有很长的路要走。我们推荐几个可能的未来研究方向。
《6.1 分布范围外的泛化和检测》
6.1 分布范围外的泛化和检测
用于训练单步或多步逆合成预测模型的反应数据集与测试分子之间的分布差距对逆合成规划性能有不可忽视的影响。正如参考文献[127]中所深入研究的那样,较小的重叠度会影响逆合成计划的性能。训练反应数据集和新分子(反应)之间较小的重叠,如文献[127]中报道的包含一般药物化学目标的阿斯利康虚拟库,将导致人工智能驱动的方法的泛化能力下降。将基于模板的方法和人工智能驱动的方法用于解决现有反应数据集分布之外的一般药物化学目标,甚至用于加工化学目标,将导致这些方法处于高风险之中。由于逆向合成的目标是合成新的分子,未来的逆向合成预测工作应强调建立模型,即使是对分布外的分子和反应也能通用,同时具有:①竞争性预测精度;②不确定性校准。这种预测不确定性的保证将使化学专家在已知分布外分子或反应具有高度不确定性的情况下进行适当干预。具有构成性的模块模型可以成为归纳到分布外样品的合格候选者。
《6.2 路线评估》
6.2 路线评估
随着计算机辅助合成计划(CASP)的发展,在评估不同CASP程序的性能时,自动路线评估变得十分迫切。除了考虑路线内每个单步反应的质量外,由逆向合成策略决定的路线的整体质量也很重要。例如,收敛合成是逆向合成策略的优先考虑,因为它减少了逆向合成途径的最大长度,提高了总体产率;保护和去保护反应类型同时出现在一条逆向合成途径中,有助于避免非选择性反应。尽管在现有文献中设计不同的启发式方法可以引导CASP程序遵循某种战略类型的逆向合成规划,但这也削弱了CASP寻找整体“更聪明”路径的潜力。未来的工作可以应用数据驱动的方法,同时也提供一个整体的路线评估,对路径级信息进行更好的编码。
《6.3 知识图谱与推理》
6.3 知识图谱与推理
知识图谱是人类知识的语义和结构化表示,它极大地促进了许多信息处理和自然语言理解问题[128‒129]。类似地,在逆合成领域,构建分子的知识图谱和连接分子的关系将有效地补充现有的非结构化和神经逆合成的算法。首先,目标分子的逆向合成路线可以通过知识图谱中查询到的最相似分子的合成路线进行规范化;其次,将化学家注释的新反应或新的合成路线纳入在线知识图谱,比将其作为更新神经逆合成算法的训练实例要有效得多。为了实现这一目标,我们认为至关重要的是:①通过合成分子的反应类型来定义分子的类别;②在这种类别的基础上制定分子之间的各种关系类别,如“Category.Share,”“ReactantOf,”和“ReagentOf;”;③通过同时考虑其他分子的个性和联系来学习分子的上下文表示,以减少噪声和模糊性。Jeong等[115]通过探索使用知识图谱来规范预测反应,已经对上述第一个研究挑战进行了合理的尝试。其余挑战——如何持续更新知识图谱和学习分子的上下文表征以促进逆向合成预测和规划——是有待进一步探索的。











 京公网安备 11010502051620号
京公网安备 11010502051620号




