
2020, Volume 6, Issue 5
Engineering >> 2020, Volume 6, Issue 5 doi: 10.1016/j.eng.2019.07.025
Nascent Polypeptide-Associated Complex Involved in the Development and Pathogenesis of Fusarium graminearum on Wheat
a State Key Laboratory for Biology of Plant Diseases and Insect Pests, Institute of Plant Protection, Chinese Academy of Agricultural Sciences, Beijing 100193, China
b College of Agriculture, Guizhou University, Guiyang 550025, China
c Department of Plant Pathology, The Ohio State University, Columbus, OH 43210, USA
# These authors contributed equally to this work.
Next Previous
Abstract
Reliable knowledge on pathogenic agents contributes to effective plant protection. For most plant pathogens, maintaining protein homeostasis (proteostasis) is essential for unfolding the cellular functions to survive and thrive. However, the fungal proteins involved in proteostasis remain poorly characterized in the process of pathogenesis. In this study, we characterized the function of the nascent polypeptide-associated complex (NAC) in Fusarium graminearum (F. graminearum, FgNAC), one of the top 10 fungal pathogens with predominant scientific/economic importance. We found that FgNACα, a subunit of FgNAC, manifests high structural and functional similarity to its homologous counterparts in yeast and other species. The mutants of F. graminearum lacking NACα are viable but suffer significant defects in vegetative growth, conidial production, and pathogenesis. In addition, we show here that FgNACα can interact with another subunit of NAC (FgNACβ) in a yeast-two-hybrid assay. The subcellular localization results show that FgNACα and FgNACβ are predominantly localized in the cytoplasm. Future studies should focus on deciphering the mechanism by which NAC orchestrates protein biogenesis and consequentially modulates development and pathogenesis.
Keywords
Fusarium head blight ; Nascent polypeptide-associated complex ; Gene knockout ; Pathogenicity ; Subcellular localization
Figures
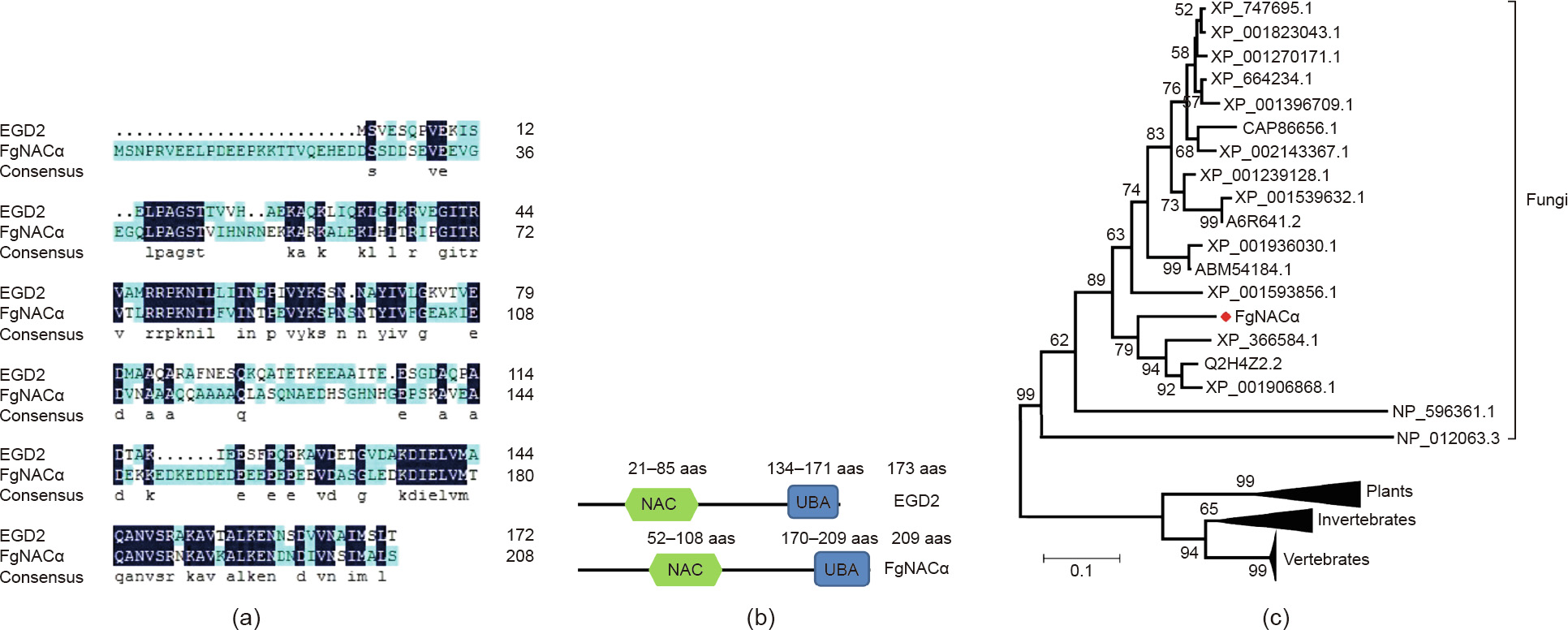
Fig. 1
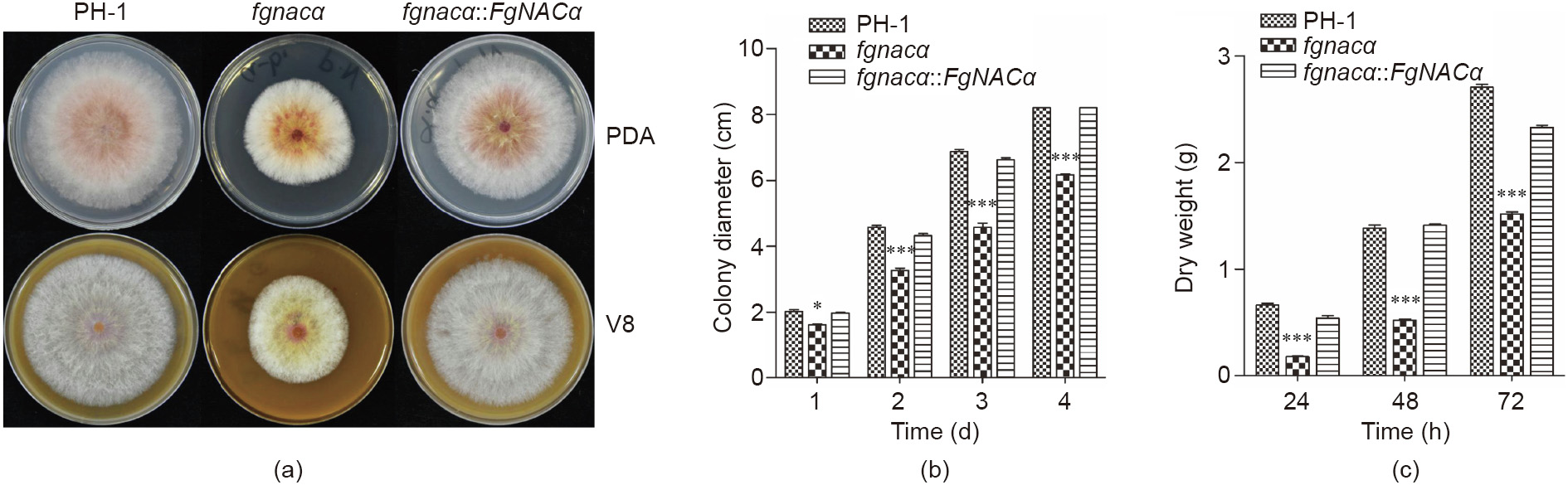
Fig. 2
Fig. 3

Fig. 4
Fig. 5
References
[ 1 ] Anderson PK, Cunningham AA, Patel NG, Morales FJ, Epstein PR, Daszak P. Emerging infectious diseases of plants: pathogen pollution, climate change and agrotechnology drivers. Trends Ecol Evol 2004;19(10):535–44. link1
[ 2 ] Fisher MC, Henk DA, Briggs CJ, Brownstein JS, Madoff LC, McCraw SL, et al. Emerging fungal threats to animal, plant and ecosystem health. Nature 2012;484(7393):186–94. link1
[ 3 ] Siersleben S, Penselin D, Wenzel C, Albert S, Knogge W. PFP1, a gene encoding an Epc-N domain-containing protein, is essential for pathogenicity of the barley pathogen Rhynchosporium commune. Eukaryot Cell 2014;13(8):1026–35. link1
[ 4 ] Kogan GL, Gvozdev VA. Multifunctional nascent polypeptide-associated complex (NAC). Mol Biol 2014;48(2):189–96. link1
[ 5 ] Beatrix B, Sakai H, Wiedmann M. The a and b subunit of the nascent polypeptide-associated complex have distinct functions. J Biol Chem 2000;275 (48):37838–45. link1
[ 6 ] Reimann B, Bradsher J, Franke J, Hartmann E, Wiedmann M, Prehn S, et al. Initial characterization of the nascent polypeptide-associated complex in yeast. Yeast 1999;15(5):397–407. link1
[ 7 ] Yang KS, Kim HS, Jin UH, Lee SS, Park JA, Lim YP, et al. Silencing of NbBTF3 results in developmental defects and disturbed gene expression in chloroplasts and mitochondria of higher plants. Planta 2007;225(6):1459–69. link1
[ 8 ] Raue U, Oellerer S, Rospert S. Association of protein biogenesis factors at the yeast ribosomal tunnel exit is affected by the translational status and nascent polypeptide sequence. J Biol Chem 2007;282(11):7809–16. link1
[ 9 ] Kirstein-Miles J, Scior A, Deuerling E, Morimoto RI. The nascent polypeptideassociated complex is a key regulator of proteostasis. EMBO J 2013;32 (10):1451–68. link1
[10] Bukau B, Deuerling E, Pfund C, Craig EA. Getting newly synthesized proteins into shape. Cell 2000;101(2):119–22. link1
[11] Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 2002;295(5561):1852–8. link1
[12] Wegrzyn RD, Deuerling E. Molecular guardians for newborn proteins: ribosome-associated chaperones and their role in protein folding. Cell Mol Life Sci 2005;62(23):2727–38. link1
[13] Lauring B, Sakai H, Kreibich G, Wiedmann M. Nascent polypeptide-associated complex protein prevents mistargeting of nascent chains to the endoplasmic reticulum. Proc Natl Acad Sci USA 1995;92(12):5411–5. link1
[14] Wiedmann B, Sakai H, Davis TA, Wiedmann M. A protein complex required for signal-sequence-specific sorting and translocation. Nature 1994;370 (6489):434–40. link1
[15] George R, Beddoe T, Landl K, Lithgow T. The yeast nascent polypeptideassociated complex initiates protein targeting to mitochondria in vivo. Proc Natl Acad Sci USA 1998;95(5):2296–301. link1
[16] Koplin A, Preissler S, Ilina Y, Koch M, Scior A, Erhardt M, et al. A dual function for chaperones SSB-RAC and the NAC nascent polypeptide-associated complex on ribosomes. J Cell Biol 2010;189(1):57–68. link1
[17] Figueroa M, Hammond-Kosack KE, Solomon PS. A review of wheat diseases—a field perspective. Mol Plant Pathol 2018;19(6):1523–36. link1
[18] Ding S, Mehrabi R, Koten C, Kang Z, Wei Y, Seong K, et al. Transducin beta-like gene FTL1 is essential for pathogenesis in Fusarium graminearum. Eukaryot Cell 2009;8(6):867–76. link1
[19] Starkey DE, Ward TJ, Aoki T, Gale LR, Kistler HC, Geiser DM, et al. Global molecular surveillance reveals novel Fusarium head blight species and trichothecene toxin diversity. Fungal Genet Biol 2007;44(11):1191–204. link1
[20] Stepien L, Chelkowski J. Fusarium head blight of wheat: pathogenic species and their mycotoxins. World Mycotoxin J 2010;3(2):107–19. link1
[21] Wang X, Cui Y, Fan F, Song Y, Ren J, Meng Q, et al. Phylogenetic, carbendazim sensitivity and mycotoxin genotype analyses of Fusarium graminearum complex species isolated from wheat Fusarium head blight in China. J Phytopathol 2010;158(7–8):576–8. link1
[22] Suga H, Kageyama K, Shimizu M, Hyakumachi M. A natural mutation involving both pathogenicity and perithecium formation in the Fusarium graminearum species complex. G3 Genes Genomes Genet 2016;6(12):3883–92. link1
[23] Park AR, Cho AR, Seo JA, Min K, Son H, Lee J, et al. Functional analyses of regulators of G protein signaling in Gibberella zeae. Fungal Genet Biol 2012;49 (7):511–20. link1
[24] Cuomo CA, Güldener U, Xu JR, Trail F, Turgeon BG, Di Pietro A, et al. The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science 2007;317(5843):1400–2. link1
[25] Wood V, Gwilliam R, Rajandream MA, Lyne M, Lyne R, Stewart A, et al. The genome sequence of Schizosaccharomyces pombe. Nature 2002;415 (6874):871–80. link1
[26] Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007;23(21):2947–8. link1
[27] Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 2016;33(7):1870–4. link1
[28] Catlett NL, Lee BN, Yoder OC, Turgeon BG. Split-marker recombination for efficient targeted deletion of fungal genes. Fungal Genet Rep 2003;50(1):9–11. link1
[29] Liu YJ, Liu X, Chen H, Zheng P, Wang W, Wang L, et al. A plastid-localized pentatricopeptide repeat protein is required for both pollen development and plant growth in rice. Sci Rep 2017;7(1):11484. link1
[30] Yuan TL, Zhang Y, Yu XJ, Cao XY, Zhang D. Optimization of transformation system of Fusarium graminearum. Plant Physiol Commun 2008;44:251–6. link1
[31] Hou ZM, Xue CY, Peng YL, Katan T, Kistler HC, Xu JR. A mitogen-activated protein kinase gene (MGV1) in Fusarium graminearum is required for female fertility, heterokaryon formation, and plant infection. Mol Plant Microbe Interact 2002;15(11):1119–27. link1
[32] Bluhm BH, Zhao X, Flaherty JE, Xu JR, Dunkle LD. RAS2 regulates growth and pathogenesis in Fusarium graminearum. Mol Plant Microbe Interact 2007;20 (6):627–36. link1
[33] Liu N, Fan F, Qiu D, Jiang L. The transcription cofactor FgSwi6 plays a role in growth and development, carbendazim sensitivity, cellulose utilization, lithium tolerance, deoxynivalenol production and virulence in the filamentous fungus Fusarium graminearum. Fungal Genet Biol 2013;58– 59:42–52. link1
[34] Zhang H, Xue C, Kong L, Li G, Xu JR. A Pmk1-interacting gene is involved in appressorium differentiation and plant infection in Magnaporthe oryzae. Eukaryot Cell 2011;10(8):1062–70. link1
[35] Sweigard JA, Chumley FG, Valent B. Disruption of a Magnaporthe grisea cutinase gene. Mol Gen Genet 1992;232(2):183–90. link1
[36] Kumar S, Stecher G, Suleski M, Hedges SB. TimeTree: a resource for timelines, timetrees, and divergence times. Mol Biol Evol 2017;34(7):1812–9. link1
[37] Ponce-Rojas JC, Avendaño-Monsalve MC, Yañez-Falcón AR, Jaimes-Miranda F, Garay E, Torres-Quiroz F, et al. abʹ-NAC cooperates with Sam37 to mediate early stages of mitochondrial protein import. FEBS J 2017;284(5):814–30. link1
[38] Lauring B, Kreibich G, Weidmann M. The intrinsic ability of ribosomes to bind to endoplasmic reticulum membranes is regulated by signal recognition particle and nascent-polypeptide-associated complex. Proc Natl Acad Sci USA 1995;92(21):9435–9. link1
[39] George R, Walsh P, Beddoe T, Lithgow T. The nascent polypeptide-associated complex (NAC) promotes interaction of ribosomes with the mitochondrial surface in vivo. FEBS Lett 2002;516(1–3):213–6. link1
[40] Ott AK, Locher L, Koch M, Deuerling E. Functional dissection of the nascent polypeptide-associated complex in Saccharomyces cerevisiae. PLoS ONE 2015;10(11):e0143457. link1
[41] Kaido M, Inoue Y, Takeda Y, Sugiyama K, Takeda A, Mori M, et al. Downregulation of the NbNACa1 gene encoding a movement-proteininteracting protein reduces cell-to-cell movement of brome mosaic virus in Nicotiana benthamiana. Mol Plant Microbe Interact 2007;20(6):671–81. link1
[42] Rospert S, Dubaquié Y, Gautschi M. Nascent-polypeptide-associated complex. Cell Mol Life Sci 2002;59(10):1632–9. link1
[43] Li X, Guo M, Xu D, Chen F, Zhang H, Pan Y, et al. The nascent-polypeptideassociated complex alpha subunit regulates the polygalacturonases expression negatively and influences the pathogenicity of Sclerotinia sclerotiorum. Mycologia 2015;107(6):1130–7. link1
[44] Li S, Peng W, Chen X, Geng X, Sun J. Identification and characterization of nascent polypeptide-associated complex alpha from Chinese mitten crab (Eriocheir sinensis): a novel stress and immune response gene in crustaceans. Fish Shellfish Immunol 2016;48:54–61. link1
[45] Li S, Chen X, Geng X, Zhan W, Sun J. Identification and expression analysis of nascent polypeptide-associated complex alpha gene in response to immune challenges in Japanese flounder Paralichthys olivaceus. Fish Shellfish Immunol 2015;46(2):261–7. link1
[46] Dahal D, Pich A, Braun HP, Wydra K. Analysis of cell wall proteins regulated in stem of susceptible and resistant tomato species after inoculation with Ralstonia solanacearum: a proteomic approach. Plant Mol Biol 2010;73 (6):643–58. link1











 京公网安备 11010502051620号
京公网安备 11010502051620号




