
2020, Volume 6, Issue 5
Engineering >> 2020, Volume 6, Issue 5 doi: 10.1016/j.eng.2020.03.004
Discovery for New Herbicide Sites of Action by Quantification of plant Primary Metabolite and Enzyme Pools
a Agricultural Biology, College of Agricultural Sciences, Colorado State University, Fort Collins, CO 80523, USA
b Natural Products Utilization Research, Agricultural Research Service, United States Department of Agriculture, University, MS 38677, USA
Next Previous
Abstract
No herbicide with a new molecular site of action (SOA) has been introduced since the 1980s. Since then, the widespread evolution of resistance of weeds to most commercial herbicides has greatly increased the need for herbicides with new SOAs. Two untried strategies for the discovery on new herbicide SOAs are discussed. Some primary metabolism intermediates are phytotoxic (e.g., protoporphyrin IX and sphingoid bases), and, because of this, the in vivo concentrations of these compounds are maintained at very low levels by plants. The determination of all primary metabolite phytotoxicities and pool sizes will identify targets of interest. Targeting SOAs that result in accumulation of phytotoxic compounds is the first novel approach to herbicide discovery. The second approach is to identify potential SOAs with very low in vivo enzyme levels. We know that higher numbers of enzyme molecules for a SOA requires more herbicide to kill a plant. Modern proteomic methods can identify low enzyme level SOAs for biorational herbicide discovery. These approaches might be useful in discovery of herbicides more closely related to natural compounds and that can be used in lower doses.
Keywords
Natural products ; Biorational discovery ; Mode of action ; Herbicide
Figures
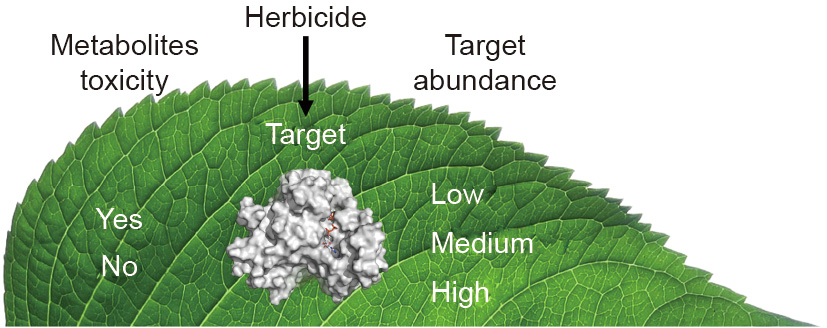
Fig. 1
References
[ 1 ] Duke SO. Why have no new herbicides modes of action appeared in recent years? Pest Manage Sci 2012;68(4):505–12. link1
[ 2 ] Heap I. The international survey of herbicide resistant weeds [Internet]. WeedScience; c1993–2020 [cited 2019 Nov 25]. Available from: www. weedscience.org. link1
[ 3 ] Copping L. The evolution of crop protection companies. Outlooks Pest Manage 2018;29(1):25–7. link1
[ 4 ] Lee DL, Prisbylla MP, Cromartie TH, Dagarin DP, Howard SW, Provan WM, et al. The discovery and structural requirements of inhibitors of p-hydroxyphenylpyruvate dioxygenase. Weed Sci 1997;45:601–9. link1
[ 5 ] Dayan FE, Duke SO, Sauldubois A, Singh N, McCurdy C, Cantrell CL. pHydroxyphenylpyruvate dioxygenase is a herbicidal target site for b-triketones from Leptospermum scoparium. Phytochemistry 2007;68(14):2004–14. link1
[ 6 ] Dayan FE, Duke SO. Natural compounds as next generation herbicides. Plant Physiol 2014;166(3):1090–105. link1
[ 7 ] Duke SO, Baerson SR, Gressel J. Genomics and weeds: a synthesis. In: Stewart CN, editor. Weed and invasive plant genomics. Ames: Blackwell Publishing; 2009. p. 221–47. link1
[ 8 ] Yan Y, Liu Q, Zang X, Yuan S, Bat-Erdene U, Nguyen C, et al. Resistance-genedirected discovery of a natural-product herbicide with a new mode of action. Nature 2018;559(7714):415–8. link1
[ 9 ] Cundliffe E. How antibiotic-producing organisms avoid suicide. Annu Rev Microbiol 1989;43(1):207–33. link1
[10] Duke SO, Stidham MA, Dayan FE. A novel genomic approach to herbicide and herbicide mode of action discovery. Pest Manage Sci 2019;75(2):314–7. link1
[11] Dayan FE, Duke SO. Protoporphyrinogen oxidase-inhibiting herbicides. In: Haye’s handbook of pesticide toxicology. San Diego: Academic Press; 2010. p. 1733–51. link1
[12] Tan S, Evans R, Singh B. Herbicidal inhibitors of amino acid biosynthesis and herbicide-tolerant crops. Amino Acids 2006;30(2):195–204. link1
[13] Amorim Franco TM, Blanchard JS. Bacterial branched-chain amino acid biosynthesis: structures, mechanisms, and drugability. Biochemistry 2017;56 (44):5849–65. link1
[14] Wittenbach VA, Abell LM. Inhibitors of valine, leucine, and isoleucine synthesis. In: Singh BK, editor. Plant amino acids: biochemistry and biotechnology. New York: Marcel Dekker; 1999. p. 385–416. link1
[15] Zhou Q, Liu W, Zhang Y, Liu KK. Action mechanisms of acetolactate synthaseinhibiting herbicides. Pestic Biochem Physiol 2007;89(2):89–96. link1
[16] Shaner DL, Singh BK. Phytotoxicity of acetohydroxyacid synthase inhibitors is not due to accumulation of 2-ketobutyrate and/or 2-aminobutyrate. Plant Physiol 1993;103(4):1221–6. link1
[17] Orcaray L, Igal M, Marino D, Zabalza A, Royuela M. The possible role of quinate in the mode of action of glyphosate and acetolactate synthase inhibitors. Pest Manage Sci 2010;66(3):262–9. link1
[18] Abbas HK, Tanaka T, Duke SO, Porter JK, Wray EM, Hodges L, et al. Fumonisinand AAL-toxin-induced disruption of sphingolipid metabolism with accumulation of free sphingoid bases. Plant Physiol 1994;106(3):1085–93. link1
[19] Tanaka T, Abbas HK, Duke SO. Structure-dependent phytotoxicity of fumonisins and related compounds in a duckweed bioassay. Phytochemistry 1993;33(4):779–85. link1
[20] Lynch DV, Dunn TM. An introduction to plant sphingolipids and are review of recent advances in understanding their metabolism and function. New Phytol 2004;161(3):677–702. link1
[21] Abbas HK, Duke SO, Shier WT, Duke MV. Inhibition of ceramide synthesis in plants by phytotoxins. In: Upadhyay RK, editor. Advances in microbial toxin research and its biotechnological exploitation. Boston: Springer; 2002. p. 211–29. link1
[22] Wang H, Li J, Bostock RM, Gilchrist DG. Apoptosis: a functional paradigm for programmed plant cell death induced by a host-selective phytotoxin and invoked during development. Plant Cell 1996;8(3):375–91. link1
[23] Foyer CH, Noctor G. Oxidant and antioxidant signaling on plants: a reevaluation of the concept of oxidative stress in a physiological context. Plant Cell Environ 2005;28(8):1056–71. link1
[24] Bernard SM, Habash DZ. The importance of cytosolic glutamine synthetase in nitrogen assimilation and recycling. New Phytol 2009;182(3):608–20. link1
[25] McNally SF, Hirel B, Gadal P, Mann AF, Stewart GR. Glutamine synthetases of higher plants: evidence for a specific isoform content related to their possible physiological role and their compartmentation within the leaf. Plant Physiol 1983;72(1):22–5. link1
[26] Edwards JW, Walker EL, Coruzzi GM. Cell-specific expression in transgenic plants reveals nonoverlapping roles for chloroplast and cytosolic glutamine synthetase. Proc Natl Acad Sci USA 1990;87(9):3459–63. link1
[27] Kamachi K, Yamaya T, Hayakawa T, Mae T, Ojima K. Vascular bundle-specific localization of cytosolic glutamine synthetase in rice leaves. Plant Physiol 1992;99(4):1481–6. link1
[28] Takano HK, Beffa R, Preston C, Westra P, Dayan FE. Reactive oxygen species trigger the fast action of glufosinate. Planta 2019;249(6):1837–49. link1
[29] Johansson L, Larsson CM. Relationship between inhibition of CO2 fixation and glutamine synthetase inactivation in Lemna gibba L. treated with L-methionine-D,L-sulphoximine (MSO). J Exp Bot 1986;37(2):221–9. link1
[30] Lu Y, Li Y, Yang Q, Zhang Z, Chen Y, Zhang S, et al. Suppression of glycolate oxidase causes glyoxylate accumulation that inhibits photosynthesis through deactivating Rubisco in rice. Physiol Plant 2014;150(3):463–76. link1
[31] Duke SO. The history and current status of glyphosate. Pest Manage Sci 2018;74(5):1027–34. link1
[32] Duke SO, Baerson SR, Rimando AM. Herbicides: glyphosate. In: Plimmer JR, editor. Encyclopedia of agrochemicals. Hobokena: John Wiley & Sons; 2003. link1
[33] Bentley R, Haslam E. The shikimate pathway—a metabolic tree with many branches. Crit Rev Biochem Mol Biol 1990;25(5):307–84. link1
[34] Shaner DL, Nadler-Hassar T, Henry WB, Koger CH. A rapid in vivo shikimate accumulation assay with excised leaf discs. Weed Sci 2005;53(6): 769–74. link1
[35] Zulet A, Zabalza A, Royuela M. Phytotoxic and metabolic effects of exogenous quinate on Pisum sativum L. J Plant Growth Regul 2013;32(4):779–88. link1
[36] Zabalza A, Orcaray L, Fernández-Escalada M, Zulet-González A, Royuela M. The pattern of shikimate pathway and phenylpropanoids after inhibition by glyphosate or quinate feeding in pea roots. Pestic Biochem Physiol 2017;141:96–102. link1
[37] Ghosh S, Chisti Y, Banerjee UC. Production of shikimic acid. Biotechnol Adv 2012;30(6):1425–31. link1
[38] Enrich LB, Scheuermann ML, Mohadjer A, Matthias KR, Eller CF, Newman MS, et al. Liquidamber styraciflua: a renewable source of shikimic acid. Tetrahedron Lett 2008;49(16):2503–5. link1
[39] Council of Europe. European pharmacopedia. 3rd ed. Strabourg: Council of Europe; 1997. link1
[40] Duke SO, Rimando AM, Duke MV, Paul RN, Ferreira JFS, Smeda RJ. Sequestration of phytotoxins by plants: implications for biosynthetic production. In: Cutler HG, Cutler SJ, editors. Biologically active natural products: agrochemicals. Boca Raton: CRC Press; 1999. p. 127–36. link1
[41] Cornish-Brown A. Why is uncompetitive inhibition so rare? A possible explanation, with implications for the design of drugs and pesticides. FEBS Lett 1986;203:25–37. link1
[42] Jessing K, Duke SO, Cedergreeen N. Potential ecological roles of artemisinin produced by Artemisia anna L. J Chem Ecol 2014;40(2):100–17. link1
[43] Vaughn SF. Glucosinolates as natural pesticides. In: Cutler HG, Cutler SJ, editors. Biologically active natural products: agrochemicals. Boca Raton: CRC Press; 1999. p. 81–91. link1
[44] Dayan FE, Rimando AM, Pan Z, Baerson SR, Gimsing AL, Duke SO. Sorgoleone. Phytochemistry 2010;71(10):1032–9. link1
[45] Díaz-Tielas C, Graña E, Maffei ME, Reigosa MJ, Sánchez-Moreiras AM. Plasma membrane depolarization precedes photosynthesis damage and long-term leaf bleaching in (E)-chalcone-treated Arabidopsis shoots. J Plant Physiol 2017;218:56–65. link1
[46] Lydon J, Duke SO. Glyphosate induction of elevated levels of hydroxybenzoic acids in higher plants. J Agric Food Chem 1988;36(4):813–8. link1
[47] Reigosa MJ, Pazos-Malvido E. Phytotoxic effects of 21 plant secondary metabolites on Arabidopsis thialiana germination and root growth. J Chem Ecol 2007;33(7):1456–66. link1
[48] Feller U, Anders I, Mae T. Rubiscolytics: fate of Rubisco after its enzymatic function in a cell is terminated. J Exp Bot 2007;59(7):1615–24. link1
[49] Barbeau WE, Kinsella JE. Ribulose bisphosphate carboxylase/oxygenase (Rubisco) from green leaves-potential as a food protein. Food Rev Int 1988;4 (1):93–127. link1
[50] Berry JA, Lorimer GH, Pierce J, Seemann JR, Meek J, Freas S. Isolation, identification, and synthesis of 2-carboxyarabinitol 1-phosphate, a diurnal regulator of ribulose bisphosphate carboxylase activity. Proc Natl Acad Sci USA 1987;84(3):734–8. link1
[51] Usuda H, Edwards GE. Inhibition of photosynthetic carbon metabolism in isolated chloroplasts by iodoacetol phosphate. Plant Physiol 1981;67(4):854–8. link1
[52] Heap I, Duke SO. Overview of glyphosate-resistant weeds worldwide. Pest Manage Sci 2018;74(5):1040–9. link1
[53] Duke SO. Enhanced metabolic degradation: the last evolved glyphosate resistance mechanism of weeds? Plant Physiol 2019;181(4):1401–3. link1
[54] Gaines TA, Zhang W, Wang D, Bukun B, Chisholm ST, Shaner DL, et al. Gene amplification confers glyphosate resistance in Amaranthus palmeri. Proc Natl Acad Sci USA 2010;107(3):1029–34. link1
[55] Culpepper AS, Grey TL, Vencill WK, Kichler JM, Webster TM, Brown SM, et al. Glyphosate-resistant Palmer amaranth (Amaranthus palmeri) confirmed in Georgia. Weed Sci 2006;54(4):620–6. link1
[56] Wiersma AT, Gaines TA, Preston C, Hamilton JP, Giacomini D, Buell CR, et al. Gene amplification of 5-enol-pyruvylshikimate-3-phosphate synthase in glyphosate-resistant Kochia scoparia. Planta 2015;241(2):463–74. link1
[57] Malone JM, Morran S, Shirley N, Boutsalis P, Preston C. EPSPS gene amplification in glyphosate-resistant Bromus diandrus. Pest Manage Sci 2016;72(1):81–8. link1
[58] Chen J, Huang H, Zhang C, Wei S, Huang Z, Chen J, et al. Mutations and amplification of EPSP gene confer resistance to glyphosate in goosegrass (Eleusine indica). Planta 2015;242(4):859–68. link1
[59] Ngo TD, Malone JM, Boutsalis P, Gill G, Preston C. ESPS gene amplification conferring resistance to glyphosate in windmill grass (Chloris truncata) in Australia. Pest Manage Sci 2018;74(5):1101–8. link1
[60] Laforest M, Soufiane B, Simard MJ, Obeid K, Page E, Nurse RE. Acetyl-CoA carboxylase overexpression in herbicide-resistant large crabgrass (Digitaria sanguinalis). Pest Manage Sci 2017;73(11):2227–35. link1











 京公网安备 11010502051620号
京公网安备 11010502051620号




