
2022, Volume 15, Issue 8
Engineering >> 2022, Volume 15, Issue 8 doi: 10.1016/j.eng.2021.06.023
Molecular Simulation of Cement-based Materials and Their Properties
a Department of Civil and Environmental Engineering, King Fahd University of Petroleum and Minerals, Dhahran 31261, Saudi Arabia
b Interdisciplinary Research Center for Construction and Building Materials, King Fahd University of Petroleum and Minerals, Dhahran 31261, Saudi Arabia
c Interdisciplinary Research Center for Advanced Materials, King Fahd University of Petroleum and Minerals, Dhahran 31261, Saudi Arabia
d Department of Chemistry, King Fahd University of Petroleum and Minerals, Dhahran 31261, Saudi Arabia
Next Previous
Abstract
Hydrated cement is one of the complex composite systems due to the presence of multi-scale phases with varying morphologies. Calcium silicate hydrate (C–S–H), which is the principal binder phase in the hydrated cement, is responsible for the stiffness, strength, and durability of Portland cement concrete. To understand the mechanical and durability behavior of concrete, it is important to investigate the interactions of hydrated cement phases with other materials at the nanoscale. In this regard, the molecular simulation of cement-based materials is an effective approach to study the properties and interactions of the cement system at the fundamental scale. Recently, many studies have been published regarding atomistic simulations to investigate the cement phases to define/explain the microscopic physical and chemical properties, thereby improving the macroscopic performance of hardened binders. The research in molecular simulation of cementitious systems involves researchers with multidisciplinary backgrounds, mainly in two areas: ① cement chemistry, where the hydration reactions govern most of the chemical and physical properties at the atomic scale; and ② computational materials science and engineering, where the bottom-up approach is required. The latter approach is still in its infancy, and as such, a study of the prevailing knowledge is useful, namely through an exhaustive literature review. This state-of-the-art report provides a comprehensive survey on studies that were conducted in this area and cites the important findings.
Keywords
Atomistic simulation ; Molecular dynamics ; Cement phases ; Hydration products ; Nanoengineering
SupplementaryMaterials
Figures

Fig. 1
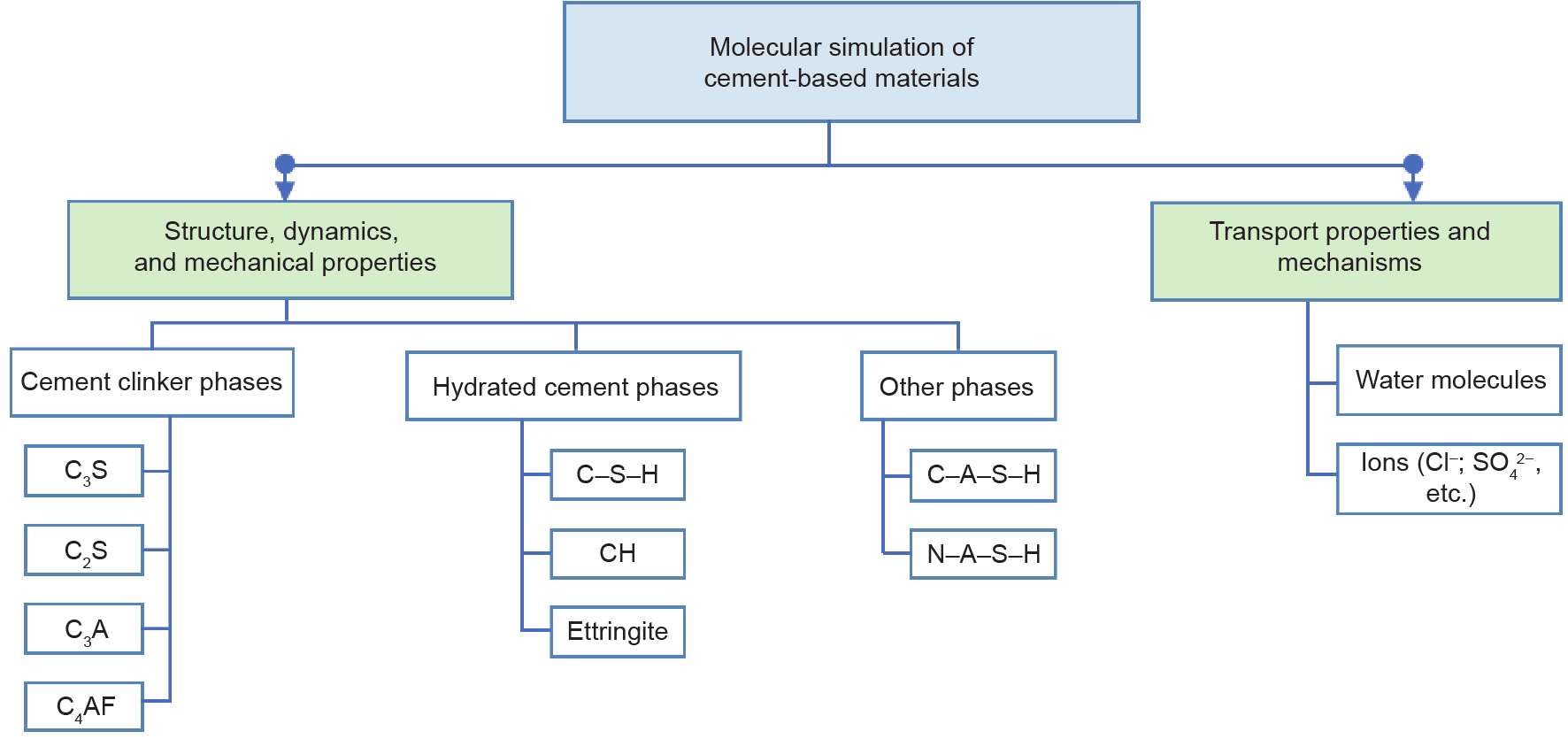
Fig. 2
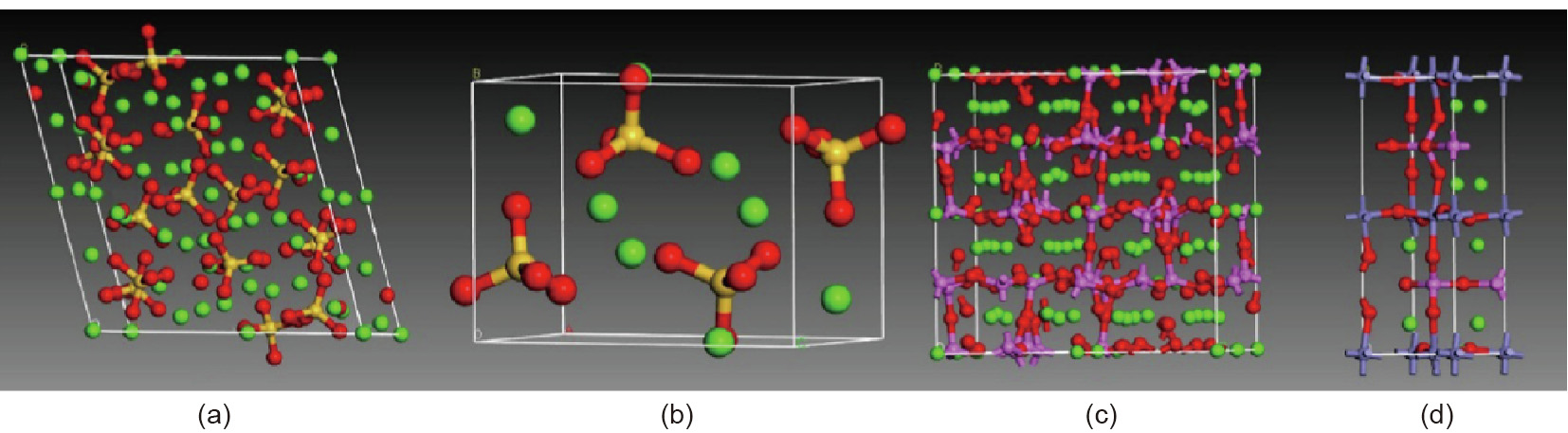
Fig. 3
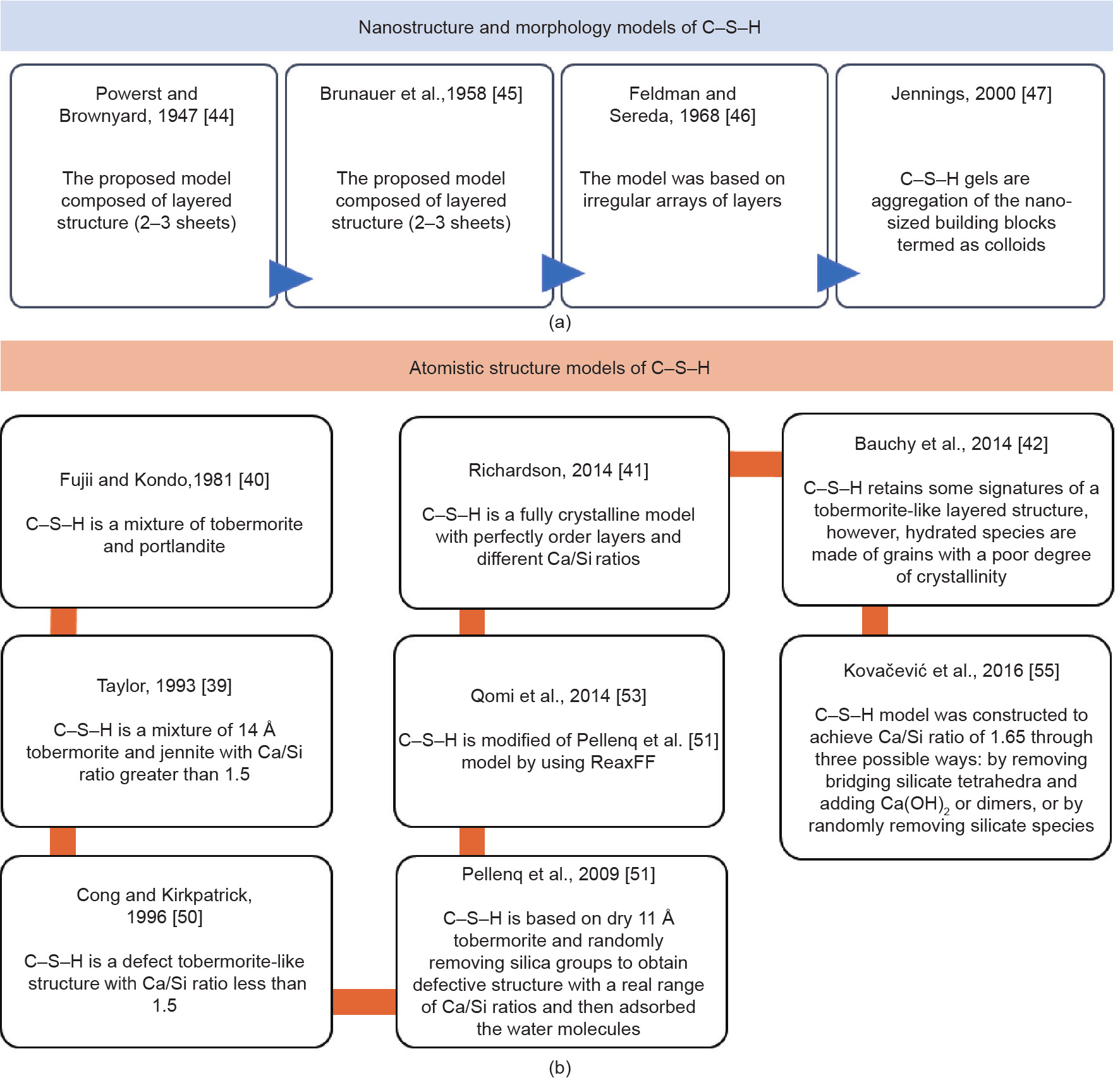
Fig. 4
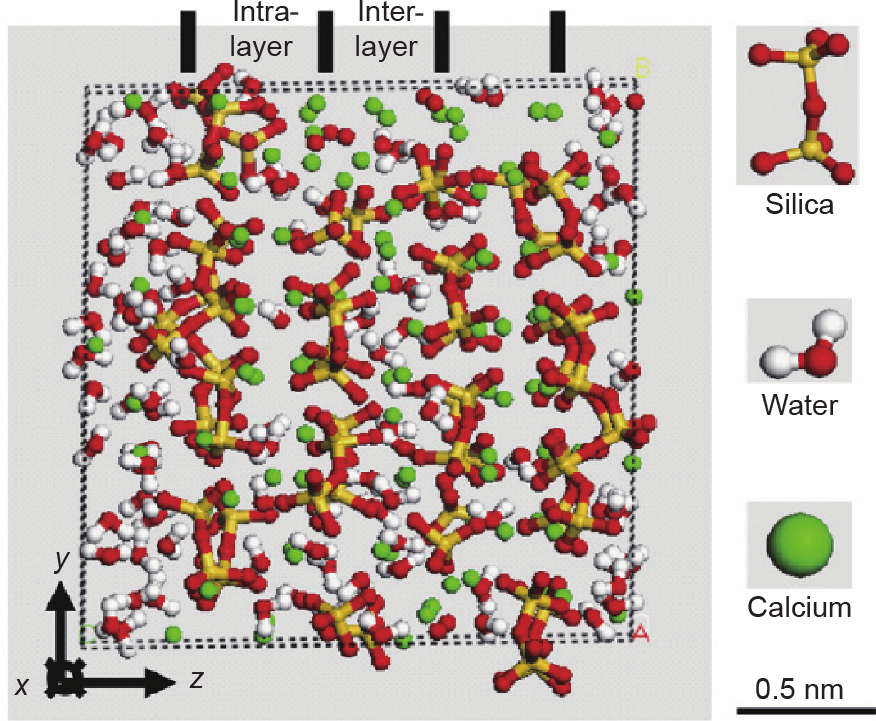
Fig. 5

Fig. 6

Fig. 7
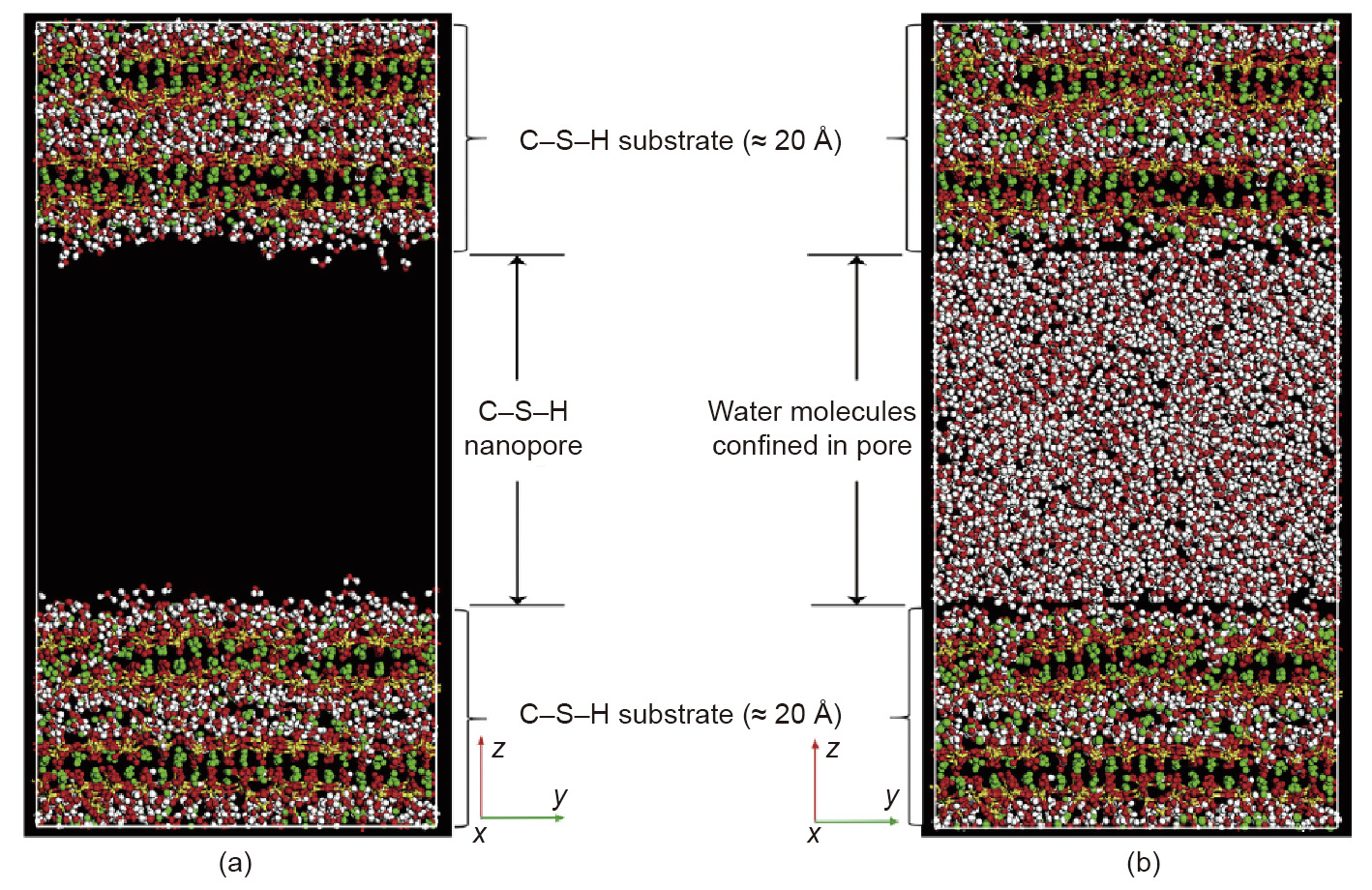
Fig. 8

Fig. 9

Fig. 10
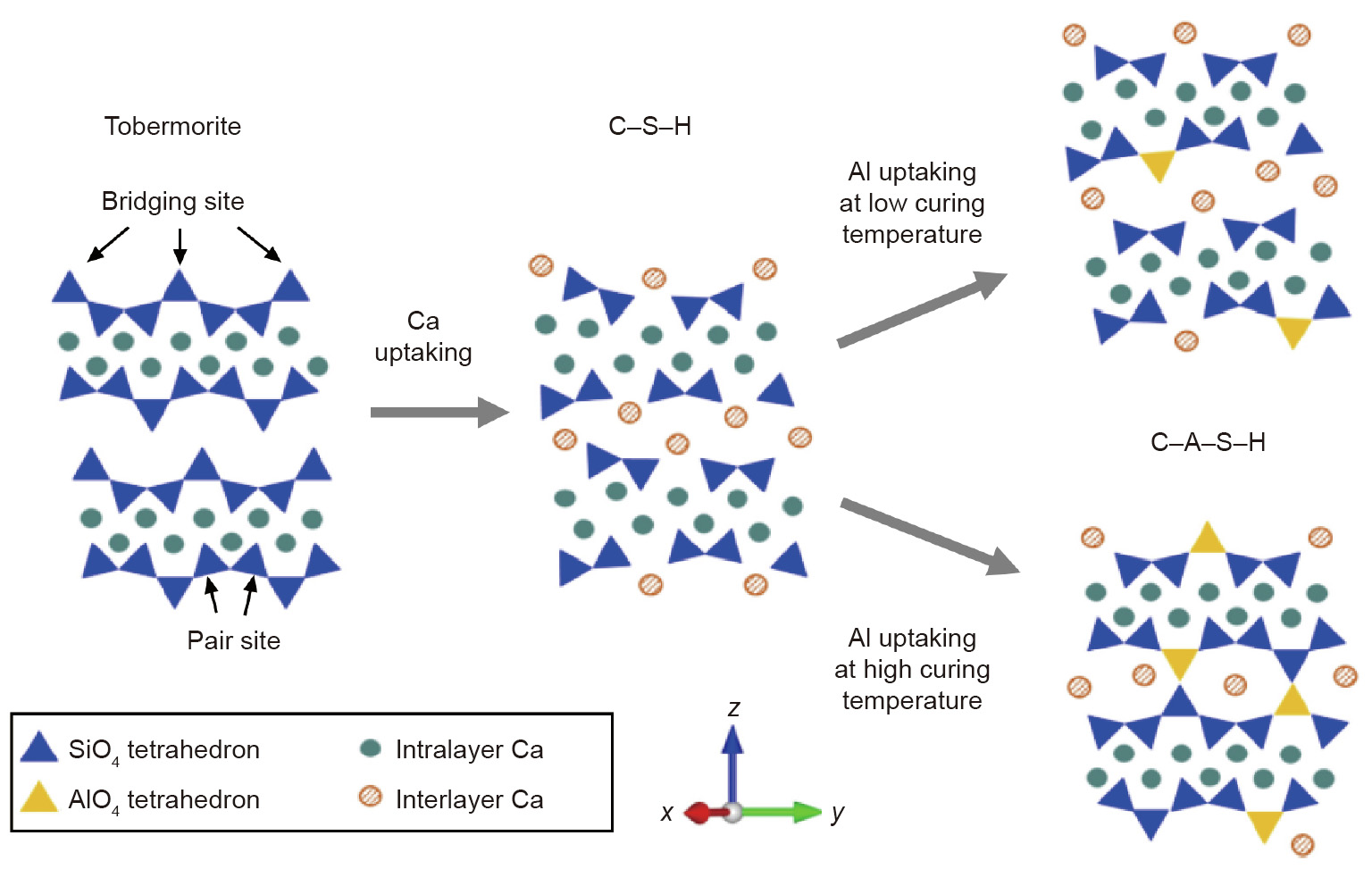
Fig. 11
References
[ 1 ] Lau D, Jian W, Yu Z, Hui D. Nano-engineering of construction materials using molecular dynamics simulations: prospects and challenges. Compos Part B Eng 2018;143:282–91. link1
[ 2 ] Sanchez F, Sobolev K. Nanotechnology in concrete—a review. Constr Build Mater 2010;24(11):2060–71. link1
[ 3 ] Beaudoin J, Odler I. Hydration, setting and hardening of Portland cement. In: Hewlett PC, Liska M, editors. Lea’s chemistry of cement and concrete. Oxford: Butterworth-Heinemann; 2019. p. 157–250. link1
[ 4 ] Al-Amoudi O, Al-Homidy A, Maslehuddin M, Saleh TA. Method and mechanisms of soil stabilization using electric arc furnace dust. Scientific reports 2017;7(1):1–10. link1
[ 5 ] Scrivener KL, Nonat A. Hydration of cementitious materials, present and future. Cement Concr Res 2011;41(7):651–65. link1
[ 6 ] Scrivener K, Ouzia A, Juilland P, Kunhi Mohamed A. Advances in understanding cement hydration mechanisms. Cement Concr Res 2019;124:105823. link1
[ 7 ] Scrivener KL, Juilland P, Monteiro PJM. Advances in understanding hydration of Portland cement. Cement Concr Res 2015;78(Pt A):38–56. link1
[ 8 ] Bullard JW, Jennings HM, Livingston RA, Nonat A, Scherer GW, Schweitzer JS, et al. Mechanisms of cement hydration. Cement Concr Res 2011;41 (12):1208–23. link1
[ 9 ] Hu Q, Aboustait M, Kim T, Ley MT, Hanan JC, Bullard J, et al. Direct threedimensional observation of the microstructure and chemistry of C3S hydration. Cement Concr Res 2016;88:157–69. link1
[10] Ahmed HR, Abduljauwad S. Significance of molecular-level behaviour incorporation in the constitutive models of expansive clays—a review. Geomech Geoengin 2018;13(2):115–38. link1
[11] Ma Z, Pathegama Gamage R, Rathnaweera T, Kong L. Review of application of molecular dynamic simulations in geological high-level radioactive waste disposal. Appl Clay Sci 2019;168:436–49. link1
[12] Abduljauwad SN, Ahmed HUR. Enhancing cancer cell adhesion with clay nanoparticles for countering metastasis. Nat Sci Reports 2019;9:1–12. link1
[13] Qu X, Wang D, Wang L, Huang Y, Hou Y, Oeser M. The state-of-the-art review on molecular dynamics simulation of asphalt binder. Adv Civ Eng 2018;2018:1–14. link1
[14] Pan J. A study of asphalt aging behavior using molecular dynamics simulations [dissertation]. Albuquerque: The University of New Mexico; 2015. link1
[15] Tam LH, Zhou A, Yu Z, Qiu Q, Lau D. Understanding the effect of temperature on the interfacial behavior of CFRP-wood composite via molecular dynamics simulations. Compos Part B Eng 2017;109:227–37. link1
[16] Zhou A, Tam LH, Yu Z, Lau D. Effect of moisture on the mechanical properties of CFRP-wood composite: an experimental and atomistic investigation. Compos Part B Eng 2015;71:63–73. link1
[17] Jin K, Qin Z, Buehler MJ. Molecular deformation mechanisms of the wood cell wall material. J Mech Behav Biomed Mater 2015;42:198–206. link1
[18] Dai W, Shui Z, Duan P. Study on the structural model of calcium silicate hydrate based on computer simulation. In: Proceedings of International Conference on Computer Technology and Science (ICCTS2012); 2012 Aug 18– 19; New Delhi, India. Singapore: IACSIT Press; 2012. p. 430–4.
[19] Akkermans RLC, Spenley NA, Robertson SH, Akkermans RLC, Spenley NA, Robertson SH. Monte Carlo methods in materials studio. Mol Simul 2013;39 (14–15):1153–64. link1
[20] Neville AM. Properties of concrete. 5th ed. San Francisco: Prentice Hall; 2011. link1
[21] Richardson I, Taylor HF. Cement chemistry. 3rd ed. London: ICE Publishing; 2017. link1
[22] Tavakoli D, Tarighat A. Molecular dynamics study on the mechanical properties of Portland cement clinker phases. Comput Mater Sci 2016;119:65–73. link1
[23] Manzano H, Durgun E, López-Arbeloa I, Grossman JC. Insight on tricalcium silicate hydration and dissolution mechanism from molecular simulations. ACS Appl Mater Interfaces 2015;7(27):14726–33. link1
[24] Mishra RK, Flatt RJ, Heinz H. Force field for tricalcium silicate and insight into nanoscale properties: cleavage, initial hydration, and adsorption of organic molecules. J Phys Chem C 2013;117(20):10417–32. link1
[25] Wu W, Al-Ostaz A, Cheng AHD, Song CR. Computation of elastic properties of Portland cement using molecular dynamics. J Nanomech Micromech 2011;1 (2):84–90. link1
[26] Wang Q, Manzano H, Guo Y, Lopez-Arbeloa I, Shen X. Hydration mechanism of reactive and passive dicalcium silicate polymorphs from molecular simulations. J Phys Chem C 2015;119(34):19869–75. link1
[27] Tao Y, Zhang W, Shang D, Xia Z, Li N, Ching W. Comprehending the occupying preference of manganese substitution in crystalline cement clinker phases: a theoretical study. Cement Concr Res 2018;109:19–29. link1
[28] Huang J, Wang B, Yu Y, Valenzano L, Bauchy M, Sant G. Electronic origin of doping-induced enhancements of reactivity: case study of tricalcium silicate. J Phys Chem C 2015;119(46):25991–9. link1
[29] Manzano H, Durgun E, Abdolhosseine Qomi MJ, Ulm FJ, Pellenq RJM, Grossman JC. Impact of chemical impurities on the crystalline cement clinker phases determined by atomistic simulations. Cryst Growth Des 2011;11(7):2964–72. link1
[30] Wang L, Hou D, Shang H, Zhao T. Molecular dynamics study on the Tri-calcium silicate hydration in sodium sulfate solution: interface structure, dynamics and dissolution mechanism. Constr Build Mater 2018;170:402–17. link1
[31] Qi C, Liu L, He J, Chen Q, Yu LJ, Liu P. Understanding cement hydration of cemented paste backfill: DFT study of water adsorption on tricalcium silicate(111) surface. Minerals 2019;9(4):202. link1
[32] Sarkar PK, Mitra N. Compressive response of tricalcium aluminate crystal: molecular dynamics investigations. Constr Build Mater 2019;224:188–97. link1
[33] Shahsavari R, Chen Lu, Tao L. Edge dislocations in dicalcium silicates: experimental observations and atomistic analysis. Cement Concr Res 2016;90:80–8. link1
[34] Sarkar PK, Mitra N. Gypsum under tensile loading: a molecular dynamics study. Constr Build Mater 2019;201:1–10. link1
[35] Roussel N, editor. Understanding the rheology of concrete. Cambridge: Woodhead Publishing; 2012. link1
[36] Richardson IG. The calcium silicate hydrates. Cement Concr Res 2008;38 (2):137–58. link1
[37] Dharmawardhana CC, Misra A, Ching WY. Quantum mechanical metric for internal cohesion in cement crystals. Sci Rep 2014;4(1):7332. link1
[38] Papatzani S, Paine K, Calabria-Holley J. A comprehensive review of the models on the nanostructure of calcium silicate hydrates. Constr Build Mater 2015;74:219–34. link1
[39] Taylor HFW. Nanostructure of C–S–H: current status. Adv Cement Base Mater 1993;1(1):38–46. link1
[40] Fujii BK, Kondo W. Heterogeneous equilibrium of calcium silicate hydrate in water. J Chem Soc, Dalton Trans 1981;30(2):645–51. link1
[41] Richardson IG. Model structures for C–(A)–S–H(I). Acta Cryst 2014;70 (6):903–23. link1
[42] Bauchy M, Qomi MJ, Ulm FJ, Pellenq RJ. Order and disorder in calcium– silicate–hydrate. J Chem Phys 2014;140(21):214503. link1
[43] Kunhi Mohamed A, Parker SC, Bowen P, Galmarini S. An atomistic building block description of C-S–H—towards a realistic C–S–H model. Cem Concr Res 2018;107:221–35. link1
[44] Powerst TC, Brownyard TL. Studies of the physical properties of hardened Portland cement paste. J Am Concr Inst 1947;43:249–336. link1
[45] Brunauer S, Kantro DL, Copeland LE. The stoichiometry of the hydration of bdicalcium silicate and tricalcium silicate at room temperature. J Am Chem Soc 1958;80(4):761–7. link1
[46] Feldman RF, Sereda PJ. A model for hydrated Portland cement paste as deduced from sorption-length change and mechanical properties. Matériaux Constr 1968;1(6):509–20. link1
[47] Jennings HM. A model for the microstructure of calcium silicate hydrate in cement paste. Cem Concr Res 2000;30(1):101–16. link1
[48] Jennings HM. Refinements to colloid model of C–S–H in cement: CM-II. Cem Concr Res 2008;38(3):275–89. link1
[49] Fujii K, Kondo W. Estimation of thermochemical data for calcium silicate hydrate (C–S–H). J Am Ceram Soc 1983;66:C220. link1
[50] Cong X, Kirkpatrick RJ. 29Si MAS NMR study of the structure of calcium silicate hydrate. Adv Cem Based Mater 1996;3(3):144–56. link1
[51] Pellenq RJ, Kushima A, Shahsavari R, Van Vliet KJ, Buehler MJ, Yip S, et al. A realistic molecular model of cement hydrates. Proc Natl Acad Sci USA 2009;106(38):16102–7. link1
[52] Hamid SA. The crystal structure of the 11 Å natural tobermorite Ca2.25 [Si3O7.5(OH)1.5]1H2O. Z Krist Cryst Mater 1981;154(1):189–98. link1
[53] Qomi MJA, Krakowiak KJ, Bauchy M, Stewart KL, Shahsavari R, Jagannathan D, et al. Combinatorial molecular optimization of cement hydrates. Nat Commun 2014;5:1–10. link1
[54] Kova G, Persson B, Nicoleau L, Nonat A, Veryazov V. Atomistic modeling of crystal structure of Ca1.67SiHx. Cement Concr Res 2015;67:197–203. link1
[55] Kovacˇevic´ G, Nicoleau L, Nonat A, Veryazov V. Revised atomistic models of the crystal structure of C–S–H with high C/S ratio. Z Phys Chem 2016;230 (9):1411–24. link1
[56] Merlino ST, Bonaccorsi EL, Armbruster TH. The real structure of tobermorite 11Å: normal and anomalous forms, OD character and polytypic modifications. Eur J Mineral. 2001;13:577–90. link1
[57] Ji Q, Pellenq RJM, Van Vliet KJ. Comparison of computational water models for simulation of calcium–silicate–hydrate. Comput Mater Sci 2012;53 (1):234–40. link1
[58] Murray SJ, Subramani VJ, Selvam RP, Hall KD. Molecular dynamics to understand the mechanical behavior of cement paste. Transp Res Rec 2010;2142(1):75–82. link1
[59] Manzano H, Ayuela A, Dolado JS. On the formation of cementitious C–S–H nanoparticles. J Comput Mater Des 2007;14(1):45–51. link1
[60] Allen AJ, Thomas JJ, Jennings HM. Composition and density of nanoscale calcium–silicate–hydrate in cement. Nat Mater 2007;6(4):311–6. link1
[61] Hou D, Ma H, Li Z. Morphology of calcium silicate hydrate (C–S–H) gel: a molecular dynamic study. Adv Cement Res 2015;27(3):135–46. link1
[62] Masoumi S, Zare S, Valipour H, Javad M, Qomi A. Effective interactions between calcium–silicate–hydrate nanolayers. J Phys Chem C 2019;123 (8):4755–66. link1
[63] Kumar A, Walder BJ, Kunhi Mohamed A, Hofstetter A, Srinivasan B, Rossini AJ, et al. The atomic-level structure of cementitious calcium silicate hydrate. J Phys Chem C 2017;121(32):17188–96. link1
[64] Mutisya SM, de Almeida JM, Miranda CR. Molecular simulations of cement based materials: a comparison between first principles and classical force field calculations. Comput Mater Sci 2017;138:392–402. link1
[65] Shahsavari R, Pellenq R, Ulm F. Empirical force fields for complex hydrated calcio-silicate layered materials. Phys Chem Chem Phys 2011;13(3):1002–11. link1
[66] Raki L, Beaudoin J, Alizadeh R, Makar J, Sato T. Cement and concrete nanoscience and nanotechnology. Materials 2010;3(2):918–42. link1
[67] Richardson IG. Tobermorite/jennite- and tobermorite/calcium hydroxide– based models for the structure of C–S–H: applicability to hardened pastes of tricalcium silicate, b-dicalcium silicate, Portland cement, and blends of Portland cement with blast-furnace slag, metakaol. Cement Concr Res 2004;34(9):1733–77. link1
[68] Hajilar S, Shafei B. Nano-scale investigation of elastic properties of hydrated cement paste constituents using molecular dynamics simulations. Comput Mater Sci 2015;101:216–26. link1
[69] Bonaccorsi E, Merlino S, Kampf AR. The crystal structure of tobermorite 14 Å (plombierite), a C–S–H phase. J Am Ceram Soc 2005;88(3):505–12. link1
[70] Bonaccorsi E, Merlino S, Taylor HFW. The crystal structure of jennite, Ca9Si6O18(OH)68H2O. Cem Concr Res 2004;34(9):1481–8. link1
[71] Hartman MR, Berliner R. Investigation of the structure of ettringite by timeof-flight neutron powder diffraction techniques. Cement Concr Res 2006;36 (2):364–70. link1
[72] Hou D, Lu Z, Zhao T, Ding Q. Reactive molecular simulation on the ordered crystal and disordered glass of the calcium silicate hydrate gel. Ceram Int 2016;42(3):4333–46. link1
[73] Chen X, Wei S, Wang Q, Tang M, Shen X, Zou X, et al. Morphology prediction of portlandite: atomistic simulations and experimental research. Appl Surf Sci 2020;502:144296. link1
[74] Tang J, Yang T, Yu C, Hou D, Liu J. Precipitated calcium hydroxide morphology in nanoparticle suspensions: an experimental and molecular dynamics study. Cement Concr Compos 2018;94:201–14. link1
[75] Laugesen JL. Density functional calculations of elastic properties of portlandite, Ca(OH)2. Cement Concr Res 2005;35(2):199–202. link1
[76] Galmarini S, Kunhi Mohamed A, Bowen P. Atomistic simulations of silicate species interaction with portlandite surfaces. J Phys Chem C 2016;120 (39):22407–13. link1
[77] Galmarini S, Bowen P. Atomistic simulation of the adsorption of calcium and hydroxyl ions onto portlandite surfaces—towards crystal growth mechanisms. Cement Concr Res 2016;81:16–23. link1
[78] Hou D, Lu Z, Zhang P, Ding Q. Molecular structure and dynamics of an aqueous sodium chloride solution in nano-pores between portlandite surfaces: a molecular dynamics study. Phys Chem Chem Phys 2016;18 (3):2059–69. link1
[79] Theodorou DN, Suter UW. Atomistic modeling of mechanical properties of polymeric glasses. Macromolecules 1986;19(1):139–54. link1
[80] Manzano H, Dolado JS, Guerrero A, Ayuela A. Mechanical properties of crystalline calcium–silicate–hydrates: comparison with cementitious C–S–H gels. Phys Status Solidi 2007;204(6):1775–80. link1
[81] Hou D, Zhao T, Jin Z, Ma H, Li Z. Molecular simulation of calcium silicate composites: structure, dynamics, and mechanical properties. J Am Ceram Soc 2015;98(3):758–69. link1
[82] Tavakoli D, Tarighat A, Beheshtian J. Nanoscale investigation of the influence of water on the elastic properties of C–S–H gel by molecular simulation. J Mater Des Appl 2017;233(7):1296–306. link1
[83] Sindu BS, Alex A, Sasmal S. Studies on structural interaction and performance of cement composite using molecular dynamics. Adv Comput Des 2018;3 (2):147–63. link1
[84] Hou D, Li H, Zhang L, Zhang J. Nano-scale mechanical properties investigation of C–S–H from hydrated tri-calcium silicate by nano-indentation and molecular dynamics simulation. Constr Build Mater 2018;189:265–75. link1
[85] Al-Ostaz A, Wu W, Cheng AHD, Song CR. A molecular dynamics and microporomechanics study on the mechanical properties of major constituents of hydrated cement. Compos Part B Eng 2010;41(7):543–9. link1
[86] Tavakoli D, Gao P, Tarighat A, Ye G. Multi-scale approach from atomistic to macro for simulation of the elastic properties of cement paste. Iran J Sci Technol Trans Civ Eng 2020;44(3):861–73. link1
[87] Manzano H, Dolado JS, Ayuela A. Elastic properties of the main species present in Portland cement pastes. Acta Mater 2009;57(5):1666–74. link1
[88] Hou D, Zhu Yu, Lu Y, Li Z. Mechanical properties of calcium silicate hydrate (C–S–H) at nano-scale: a molecular dynamics study. Mater Chem Phys 2014;146(3):503–11. link1
[89] Hou D, Zhang J, Li Z, Zhu Yu. Uniaxial tension study of calcium silicate hydrate (C–S–H): structure, dynamics and mechanical properties. Mater Struct Constr 2015;48(11):3811–24. link1
[90] Hou D, Zhao T, Wang P, Li Z, Zhang J. Molecular dynamics study on the mode I fracture of calcium silicate hydrate under tensile loading. Eng Fract Mech 2014;131:557–69. link1
[91] Hou D, Yu J, Jin Z, Hanif A. Molecular dynamics study on calcium silicate hydrate subjected to tension loading and water attack: structural evolution, dynamics degradation and reactivity mechanism. Phys Chem Chem Phys 2018;20(16):11130–44. link1
[92] Zaoui A. Insight into elastic behavior of calcium silicate hydrated oxide (C–S– H) under pressure and composition effect. Cement Concr Res 2012;42 (2):306–12. link1
[93] Wang XF, Li TR, Wei P, Li DW, Han NX, Xing F, et al. Computational study of the nanoscale mechanical properties of C–S–H composites under different temperatures. Comput Mater Sci 2018;146:42–53. link1
[94] Honorio T. Monte Carlo molecular modeling of temperature and pressure effects on the interactions between crystalline calcium silicate hydrate layers. Langmuir 2019;35(11):3907–16. link1
[95] Hou D, Ma H, Zhu Y, Li Z. Calcium silicate hydrate from dry to saturated state: structure, dynamics and mechanical properties. Acta Mater 2014;67:81–94. link1
[96] Lin W, Zhang C, Fu J, Xin H. Dynamic mechanical behaviors of calcium silicate hydrate under shock compression loading using molecular dynamics simulation. J Non-Cryst Solids 2018;500:482–6. link1
[97] Fan D, Yang S. Mechanical properties of C–S–H globules and interfaces by molecular dynamics simulation. Constr Build Mater 2018;176:573–82. link1
[98] Fu J, Bernard F, Kamali-Bernard S. Assessment of the elastic properties of amorphous calcium silicates hydrates (I) and (II) structures by molecular dynamics simulation. Mol Simul 2018;44(4):285–99. link1
[99] Rivas Murillo JS, Mohamed A, Hodo W, Mohan RV, Rajendran A, Valisetty R. Computational modeling of shear deformation and failure of nanoscale hydrated calcium silicate hydrate in cement paste: calcium silicate hydrate Jennite. Int J Damage Mech 2016;25(1):98–114. link1
[100] Manzano H, Duque-Redondo E, Masoero E, López-Arbeloa I. The role of water on C–S–H gel shear strength studied by molecular dynamics simulations. In: Hellmich C, Pichler B, Kollegger J, editors. CONCREEP 10: mechanics and physics of creep, shrinkage, and durability of concrete and concrete structures; 2015. p. 899–907.
[101] Palkovic SD, Yip S, Büyüköztürk O. Constitutive response of calcium–silicate– hydrate layers under combined loading. J Am Ceram Soc 2017;100 (2):713–23. link1
[102] Rivas Murillo JS, Hodo W, Mohamed A, Mohan RV, Rajendran A, Valisetty R. A molecular dynamics investigation of hydrostatic compression characteristics of mineral Jennite. Cem Concr Res 2017;99:62–9. link1
[103] Espinosa IMP, Hodo W, Rivas Murillo JS, Rajendran AM, Mohan RV. Constitutive stiffness characteristics of cement paste as a multiphase composite system—a molecular dynamics-based model. J Eng Mater Technol 2017;139(4):041007. link1
[104] Bauchy M, Laubie H, Abdolhosseini Qomi MJ, Hoover CG, Ulm FJ, Pellenq RJM. Fracture toughness of calcium–silicate–hydrate from molecular dynamics simulations. J Non-Cryst Solids 2015;419:58–64. link1
[105] Jalilvand S, Shahsavari R. Molecular mechanistic origin of nanoscale contact, friction, and scratch in complex particulate systems. ACS Appl Mater Interfaces 2015;7(5):3362–72. link1
[106] Hajilar S, Shafei B. Mechanical failure mechanisms of hydrated products of tricalcium aluminate: a reactive molecular dynamics study. Mater Des 2016;90:165–76. link1
[107] Sarkar PK, Mitra N, Prasad D. Molecular level deformation mechanism of ettringite. Cement Concr Res 2019;124:105836. link1
[108] Li D, Zhao W, Hou D, Zhao T. Molecular dynamics study on the chemical bound, physical adsorbed and ultra-confined water molecules in the nanopore of calcium silicate hydrate. Constr Build Mater 2017;151:563–74. link1
[109] Manzano H, Moeini S, Marinelli F, van Duin ACT, Ulm F-J, Pellenq R-M. Confined water dissociation in microporous defective silicates: mechanism, dipole distribution, and impact on substrate properties. J Am Chem Soc 2012;134(4):2208–15. link1
[110] Hou D, Li Z, Zhao T, Zhang P. Water transport in the nano-pore of the calcium silicate phase: reactivity, structure and dynamics. Phys Chem Chem Phys 2015;17(2):1411–23. link1
[111] Hou D, Zhao T, Ma H, Li Z. Reactive molecular simulation on water confined in the nanopores of the calcium silicate hydrate gel: structure, reactivity, and mechanical properties. J Phys Chem C 2015;119(3):1346–58. link1
[112] Hou D, Li D, Zhao T, Li Z. Confined water dissociation in disordered silicate nanometer-channels at elevated temperatures: mechanism, dynamics and impact on substrates. Langmuir 2016;32(17):4153–68. link1
[113] Yoon S, Monteiro PJM. Molecular dynamics study of water molecules in interlayer of 14 Å tobermorite. J Adv Concr Technol 2013;11(6):180–8. link1
[114] Hou D, Ma H, Li Z, Jin Z. Molecular simulation of ‘‘hydrolytic weakening”: a case study on silica. Acta Mater 2014;80:264–77. link1
[115] Tang S, A H, Chen J, Yu W, Yu P, Chen E, et al. The interactions between water molecules and C–S–H surfaces in loads-induced nanopores: a molecular dynamics study. Appl Surf Sci 2019;496:143744.
[116] Wang P, Jia Y, Li T, Hou D, Zheng Q. Molecular dynamics study on ions and water confined in the nanometer channel of Friedel’s salt: structure, dynamics and interfacial interaction. Phys Chem Chem Phys 2018;20 (42):27049–58. link1
[117] Zhang P, Hou D, Liu Q, Liu Z, Yu J. Water and chloride ions migration in porous cementitious materials: an experimental and molecular dynamics investigation. Cement Concr Res 2017;102:161–74. link1
[118] Honorio T, Benboudjema F, Bore T, Ferhat M, Vourc’h E. The pore solution of cement-based materials: structure and dynamics of water and ions from molecular simulations. Phys Chem Chem Phys 2019;21(21):11111–21. link1
[119] Hou D, Li Z. Molecular dynamics study of water and ions transported during the nanopore calcium silicate phase: case study of jennite. J Mater Civ Eng 2014;26(5):930–40. link1
[120] Hou D, Li Z. Molecular dynamics study of water and ions transport in nanopore of layered structure: a case study of tobermorite. Microporous Mesoporous Mater 2014;195:9–20. link1
[121] Hou D, Jia Y, Yu J, Wang P, Liu Q. Transport properties of sulfate and chloride ions confined between calcium silicate hydrate surfaces: a molecular dynamics study. J Phys Chem C 2018;122(49):28021–32. link1
[122] Zhou Y, Hou D, Jiang J, Wang P. Chloride ions transport and adsorption in the nano-pores of silicate calcium hydrate: experimental and molecular dynamics studies. Constr Build Mater 2016;126:991–1001. link1
[123] Hou D, Hu C, Li Z. Molecular simulation of the ions ultraconfined in the nanometer- channel of calcium silicate hydrate: hydration mechanism, dynamic properties, and influence on the cohesive strength. Inorg Chem 2017;56(4):1881–96. link1
[124] Hou D, Li D, Yu J, Zhang P. Insights on capillary adsorption of aqueous sodium chloride solution in the nanometer calcium silicate channel: a molecular dynamics study. J Phys Chem C 2017;121(25):13786–97. link1
[125] Yang J, Jia Y, Hou D, Wang P, Jin Z, Shang H, et al. Na and Cl immobilization by size controlled calcium silicate hydrate nanometer pores. Constr Build Mater 2019;202:622–35. link1
[126] Yang J, Hou D, Ding Q. Ionic hydration structure, dynamics and adsorption mechanism of sulfate and sodium ions in the surface of calcium silicate hydrate gel: a molecular dynamics study. Appl Surf Sci 2018;448:559–70. link1
[127] Zhou Y, Hou D, Jiang J, Liu L, She W, Yu J. Experimental and molecular dynamics studies on the transport and adsorption of chloride ions in the nano-pores of calcium silicate phase: the influence of calcium to silicate ratios. Microporous Mesoporous Mater 2018;255:23–35. link1
[128] Dufresne A, Arayro J, Zhou T, Ioannidou K, Ulm FJ, Pellenq R, et al. Atomistic and mesoscale simulation of sodium and potassium adsorption in cement paste. J Chem Phys 2018;149(7):074705. link1
[129] Zehtab B, Tarighat A. Molecular dynamics simulation to assess the effect of temperature on diffusion coefficients of different ions and water molecules in C–S–H. Mech Time-Depend Mater 2018;22(4):483–97. link1
[130] Hou D, Zhang Q, Xu X, Zhang J, Li W, Wang P. Insights on ions migration in the nanometer channel of calcium silicate hydrate under external electric field. Electrochim Acta 2019;320:134637. link1
[131] Kalinichev AG, Kirkpatrick RJ. Molecular dynamics modeling of chloride binding to the surfaces of calcium hydroxide, hydrated calcium aluminate, and calcium silicate phases. Chem Mater 2002;14(8):3539–49. link1
[132] Hajilar S, Shafei B. Structure, orientation, and dynamics of water-soluble ions adsorbed to basal surfaces of calcium monosulfoaluminate hydrates. Phys Chem Chem Phys 2018;20(38):24681–94. link1
[133] Huang X, Hu S, Wang F, Yang L, Rao M, Tao Y. Enhanced sulfate resistance: the importance of iron in aluminate hydrates. ACS Sustain Chem Eng 2019;7 (7):6792–801. link1
[134] Youssef M, Pellenq RM, Yildiz B. Docking 90Sr radionuclide in cement: an atomistic modeling study. Phys Chem Earth 2014;70-71:39–44. link1
[135] Bu J, Gonzalez Teresa R, Brown KG, Sanchez F. Adsorption mechanisms of cesium at calcium–silicate–hydrate surfaces using molecular dynamics simulations. J Nucl Mater 2019;515:35–51. link1
[136] Jiang J, Zheng Q, Yan Y, Guo D, Wang F, Wu S, et al. Design of a novel nanocomposite with C–S–H@LA for thermal energy storage: a theoretical and experimental study. Appl Energy 2018;220:395–407. link1
[137] Shi C, Qu B, Provis JL. Recent progress in low-carbon binders. Cement Concr Res 2019;122:227–50. link1
[138] Lolli F, Manzano H, Provis JL, Bignozzi MC, Masoero E. Atomistic simulations of geopolymer models: the impact of disorder on structure and mechanics. ACS Appl Mater Interfaces 2018;10(26):22809–20. link1
[139] Sadat MR, Bringuier S, Asaduzzaman A, Muralidharan K, Zhang L. A molecular dynamics study of the role of molecular water on the structure and mechanics of amorphous geopolymer binders. J Chem Phys 2016;145 (13):134706. link1
[140] Bagheri A, Nazari A, Sanjayan JG, Duan W. Molecular simulation of water and chloride ion diffusion in nanopores of alkali-activated aluminosilicate structures. Ceram Int 2018;44(17):20723–31. link1
[141] Lyngdoh GA, Kumar R, Krishnan NMA, Das S. Realistic atomic structure of fly ash-based geopolymer gels: insights from molecular dynamics simulations. J Chem Phys 2019;151(6):064307. link1
[142] Hou D, Zhang J, Pan W, Zhang Y, Zhang Z. Nanoscale mechanism of ions immobilized by the geopolymer: a molecular dynamics study. J Nucl Mater 2020;528:151841. link1
[143] Hou D, Zhang Y, Yang T, Zhang J, Pei H, Zhang J, et al. Molecular structure, dynamics, and mechanical behavior of sodium aluminosilicate hydrate (NASH) gel at elevated temperature: a molecular dynamics study. Phys Chem Chem Phys 2018;20(31):20695–711. link1
[144] Zhang Y, Zhang J, Jiang J, Hou D, Zhang J. The effect of water molecules on the structure, dynamics, and mechanical properties of sodium aluminosilicate hydrate (NASH) gel: a molecular dynamics study. Constr Build Mater 2018;193:491–500. link1
[145] Puertas F, Palacios M, Manzano H, Dolado JS, Rico A, Rodríguez J. A model for the C–A–S–H gel formed in alkali-activated slag cements. J Eur Ceram Soc 2011;31(12):2043–56. link1
[146] Geng G, Vasin RN, Li J, Qomi MJA, Yan J, Wenk HR, et al. Preferred orientation of calcium aluminosilicate hydrate induced by confined compression. Cement Concr Res 2018;113:186–96. link1
[147] Yang J, Hou D, Ding Q. Structure, dynamics, and mechanical properties of cross-linked calcium aluminosilicate hydrate: a molecular dynamics study. ACS Sustain Chem Eng 2018;6(7):9403–17. link1
[148] Wan X, Hou D, Zhao T, Wang L. Insights on molecular structure and microproperties of alkali-activated slag materials: a reactive molecular dynamics study. Constr Build Mater 2017;139:430–7. link1
[149] Zhang J, Yang J, Hou D, Ding Q. Molecular dynamics study on calcium aluminosilicate hydrate at elevated temperatures: structure, dynamics and mechanical properties. Mater Chem Phys 2019;233:276–87. link1
[150] Hou D, Li T, Wang P. Molecular dynamics study on the structure and dynamics of NaCl solution transport in the nanometer channel of CASH gel. ACS Sustain Chem Eng 2018;6:9498–509. 151. link1
[151] Hou D, Li T. Influence of aluminates on the structure and dynamics of water and ions in the nanometer channel of calcium silicate hydrate (C–S–H) gel. Phys Chem Chem Phys 2018;20(4):2373–87. link1
[152] Ding Q, Yang J, Hou D, Zhang G. Insight on the mechanism of sulfate attacking on the cement paste with granulated blast furnace slag: an experimental and molecular dynamics study. Constr Build Mater 2018;169:601–11. link1
[153] Zhang G, Zhang X, Ding Q, Hou D, Liu K. Microstructural evolution mechanism of C–(A)–S–H gel in Portland cement pastes affected by sulfate ions. J Wuhan Univ Technol Mater Sci 2018;33(3):639–47. link1
[154] Hasami M, Tarighat A. Proposing new pozzolanic activity index based on water adsorption energy via molecular dynamic simulations. Constr Build Mater 2019;213:492–504. link1











 京公网安备 11010502051620号
京公网安备 11010502051620号




