

《1. 引言》
1. 引言
人源诺如病毒(human norovirus, HuNoV)是非细菌性胃肠炎的主要病原,导致全世界50%不同年龄段人群发病[1,2]。诺如病毒(norovirus, NoV)是类属于杯状病毒科(Caliciviridae)的无包膜单股正链RNA病毒,基因组长度约为7.5 kb [3]。基因组编码3个开放阅读框(open reading frame,  ,
, 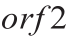 ,
, 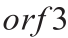 )。编码包括RNA依赖性RNA聚合酶(RNA-dependent RNApolymerase, RdRp)在内的非结构蛋白,编码主要结构蛋白(VP1),编码次要结构蛋白(VP2)[4]。NoV分为10个基因族(GI-GX),包含40余种基因型,每个基因型又含有多种亚型[5]。VP1氨基酸序列差异小于14.1%的病毒株被认为是同一基因亚型水平,存在14.3%~43.8%差异的被认为属于同一聚类水平(基因型),差异为44.9%~61.4%的属于同一族水平[6]。GI和GII族HuNoV主要引起人类急性胃肠炎,其中,过去的几十年中GII.4型HuNoV一直是流行毒株[7]。
)。编码包括RNA依赖性RNA聚合酶(RNA-dependent RNApolymerase, RdRp)在内的非结构蛋白,编码主要结构蛋白(VP1),编码次要结构蛋白(VP2)[4]。NoV分为10个基因族(GI-GX),包含40余种基因型,每个基因型又含有多种亚型[5]。VP1氨基酸序列差异小于14.1%的病毒株被认为是同一基因亚型水平,存在14.3%~43.8%差异的被认为属于同一聚类水平(基因型),差异为44.9%~61.4%的属于同一族水平[6]。GI和GII族HuNoV主要引起人类急性胃肠炎,其中,过去的几十年中GII.4型HuNoV一直是流行毒株[7]。
由于缺乏简便可靠的体外培养体系,目前常用于检测HuNoV的方法包括电子显微镜法(electron microscopy, EM)[8]、酶联免疫吸附试验(enzyme-linked immunosorbent assay, ELISA)[9]以及诸如RT-PCR和RT-qPCR的分子方法等[10–12]。其中,RT-qPCR方法由于无需使用琼脂糖凝胶分析、可即时确认、特异性和灵敏度高于RT-PCR等特点被广泛应用,成为HuNoV检测的“金标准”。多年来,-连接处被认为是HuNoV基因组中最保守的区域,并以此设计了引物探针用于RT-qPCR检测[13–15]。2003年,Kageyama等[13]设计了第一套用于检测GI和GII族NoV的引物和探针;2004—2007年,引物和探针又得到了更新[13,16–19]。目前,这些引物和探针仍然被广泛用于HuNoV的检测中,亦未进行更新。NoV基因组由RNA编码,易突变,从而导致设计的引物和(或)探针无法有效地与已进化的新病毒核酸进行匹配。数据库中公布了诸多的新病毒株基因组信息,有必要利用更新的序列数据库重新设计新的引物/探针组,从而优化HuNoV分子检测体系。
本研究从GenBank数据库下载了2010年1月到2018年5月的GI和GII族HuNoV基因组序列,进行了生物信息学分析后,在一组具有HuNoV典型基因型的基因组序列中,重新设计和评估GI和GII族HuNoV的引物/探针,建立相应的RT-qPCR检测体系,并与现有Kageyama所报道的检测体系进行了比较。
《2. 材料与方法》
2. 材料与方法
《2.1. 目标序列的收集》
2.1. 目标序列的收集
从美国国家生物技术信息中心(National Center for Biotechnology Information, NCBI)† 检 索自2010年1月到2018年5 月来源于中国的GII族HuNoV完整基因组132条。鉴于国内鲜有2010年1月之后完整GI族HuNoV基因组数据,因此本研究从NCBI下载了57条完整GI族HuNoV基因组序列,并根据2.2节中提到的方法进行比对,以确定GI的保守区域。使用GI.2序列较为保守的片段(GenBank号:KF306212.1)5288~5427作为基本局部比对搜索工具(Basic Local Alignment Search Tool, BLSAT)搜索的模板,以相似性高于80%为标准,从NCBI中检索到了2010年1月后公布的90条GI族上述区域的核酸片段。
† https://www.ncbi.nlm.nih.gov/
《2.2. 序列的多重比对和引物探针的优化设计》
2.2. 序列的多重比对和引物探针的优化设计
对2.1节下载的GI族(57条 ) 和GII族(132条 )NoV的全基因组序列分别以Simplot程序(3.5.1版)[20]分析各自的保守区域,用于后续设计新的引物和探针。 本研究以GenBank号为KF306212.1 (GI.2)和KU870455.1 (GII.6)的全基因组序列为参考序列,利用MEGA 7.0软件 ‡ 分别对GI和GII族HuNoV基因组保守区序列构建最大似然(maximum likelihood, ML)系统发育树。使用bootstrap测试(1000次计算)评估集群结果的可靠性。在分析GI和GII族HuNoV核苷酸(nt)保守区及保守趋势分布后,结合引物和探针设计的基本规则,设计两套引物/探针(表1),并命名为LZIF、LZIR-A、LZIR-B、LZIP(GI族)及LZIIF-A、LZIIF-B、LZIIR和LZIIP(GII族)。
‡ https://www.megasoftware.net/
《表1》
表1 本研究中新的HuNoV检测引物和探针相关信息

FAM: 6-carboxy-fluorescein;HEX: 5-hexachloro-fluorescein;MGB: minor groove binder.
《2.3. 从临床样本中提取病毒 RNA》
2.3. 从临床样本中提取病毒 RNA
HuNoV阳性临床样本由北京市疾病预防控制中心高志勇博士、浙江省疾病预防控制中心廖宁波博士和中国疾病预防控制中心李慧颖博士提供。以磷酸盐缓冲液(phosphate-buffered saline, PBS, pH=7.2~7.4)按照10%(m/V)稀释粪便样品,以4378 r·min−1 离心5 min,上清分装后于−80 ℃保存。取140 μL处理样本,按照HiPure病毒RNA抽提试剂盒(广州美基生物科技有限公司)说明书提取核酸,最后使用60 μL不含RNase的ddH2O进行RNA洗脱;立即使用或分装后储存于−80 ℃冰箱备用。
《2.4. 制备 RNA 体外转录反应模板》
2.4. 制备 RNA 体外转录反应模板
以GI.5型HuNoV(样品57565)为模板,引物GI-4610-F (5'-TGATGCWGAYTATACAGCWTGGGA -3')和GI-5704R (5'-CATYTTYCCAACCCARCCATTATA-CAT-3')逆转录扩增目标片段;以GII.17型HuNoV(样品15651202)的RNA为模板,以引物P290 [21]和GIISKR[22]逆转录扩增目标片段。GI族的目标片段位置为4558~5675(以GenBank号KF306212.1基因组为计算依据),GII族的扩增片段位置为4298~5383(GenBank号KU870455.1基因组为计算依据)。采用一步RT-PCR试剂盒(Takara,中国大连)说明书的方案,在总体积为20.0 μL的条件下进行RT-PCR。PCR产物克隆到pMD18-T载体(Takara,中国大连)中,并转化到大肠杆菌TOP10(GENEWIZ,中国苏州)。测序验证后,以核酸内切酶Hin d III和Bam H I对重组质粒进行双酶切,将目标片段分别插入到含有T7启动子的pET-28a (+)质粒(ThermoFisher,中国上海)中,获得重组质粒pET28-GI-M和pET28-GII-M并送往GENEWIZ(中国苏州)测序。
随后参照T7 RNA聚合酶(Beyotime,中国上海)说明书,进行重组质粒中目标DNA片段体外转录,获得RNA模板。纯化后的RNA分装并于−80 ℃保存。
《2.5. 一步法 RT-qPCR 实验》
2.5. 一步法 RT-qPCR 实验
以一步法 PrimeScriptTM RT-PCR kit (Perfect Real Time)试剂盒(Takara,中国大连),在CFX96型荧光定量PCR仪(Biorad,美国)中进行新型双重RT-qPCR(ND-RT-qPCR)反应。使用改良的荧光基团和猝灭剂合成了重新设计的探针(GENEWIZ,中国苏州)。每10.0 μL反应体系中,包含5.0 μL 2×一步法RT-PCR缓冲液III、0.2 μL Ex Taq HS(5 U·μL −1 ,1 U= 1 μmol·min–1 )、0.2 μL PrimeScript RT酶混合物II、1.2 μL不含RNase的水、1.0 μL混合引物(每条引物的终浓度为0.8 μmol·L –1 )、0.2 μL GI族探针(10.0 μmol·L –1 )、0.2 μL GII族探针(10.0 μmol·L –1 )和2.0 μL 模板RNA。循环参数如下:42 ℃逆转录3 min;95 ℃热裂解5 s;然后进行40个循环反应,包括95 ℃ 3 s,60 ℃ 7 s,在每个延伸步骤结束时读取荧光值。阈值使用Bio-Rad CFX96管理软件默认参数确定。
《2.6. 基于模板 RNA 的 RT-qPCR 标准曲线》
2.6. 基于模板 RNA 的 RT-qPCR 标准曲线
使用NanoDrop 2000C分光光度计(NanoDrop Technologies Inc., Wilmington, DE, USA)测量2.4节中制备的体外转录病毒RNA模板的浓度,并转换为基因组拷贝数。RNA拷贝总数计算如下:拷贝数=RNA浓度(ng·µL −1 )/摩尔质量×6.02×1023 。RNA模板进行10倍系列稀释(100 ~106 拷贝·µL −1 )。绘制RNA拷贝数与荧光信号达到阈值所需循环数(定量循环,quantification cycle, Cq)的关系,获得GI族和GII族NoV的定量标准曲线。重复此实验并计算平均Cq值,以确定标准曲线的斜率和回归值,R 2 系数通过Origin 2017软件进行拟合计算获得。随后,使用以下公式计算PCR反应效率:效率E = 10–1/斜率 –1 [23]。
《2.7. 三种 RT-qPCR 方法检测 HuNoV 的评价》
2.7. 三种 RT-qPCR 方法检测 HuNoV 的评价
以Kageyama等设计的HuNoV荧光定量检测方法为对照,与本研究建立的ND-RT-qPCR(2.3节)进行比较分析。
实验A中,按照2.5节中所述进行ND-RT-qPCR。实验B中,除使用Kageyama等[13]设计的引物和探针外,其他条件与实验A相同。实验C中,使用Kageyama等的RT-qPCR反应体系,反应程序按照参考文献[24]进行设置。RT-qPCR反应混合物包括:5.0 μL 2×一步法RT-PCR Buffer III、0.2 μL Ex Taq HS (5 U·μL −1 )、0.2 μL PrimeScript RT酶混合物II、每条引物0.4 μL(COGIF和COGIR,或COGIIF和COGIIR,浓度为 10 μmol·L −1 )、0.2 μL每 条 探 针[RING1(a)-TP、RING1(b)-TP或RING2-TP,浓度为10 μmol·L −1 ]、2.0 μL RNA样本,以不含RNase的ddH2O调整总体积为10.0 μL。热循环参数为:42 ℃ 30 min;95 ℃ 5 min;95 ℃ 15 s,56 ℃ 1 min,40次循环,在每个56 ℃延伸步骤结束时读取荧光值。
以Cq值为40作为阴性样品的阈值。比较上述A、B和C三种不同RT-qPCR反应体系获得同一个样本的Cq值。
《3. 结果》
3. 结果
《3.1. 序列保守性分析》
3.1. 序列保守性分析
利用PlotSimilarity方法确定了HuNoV检测的候选保守区。本研究共分析了57条GI族和132条GII族HuNoV基因组序列(涵盖国内HuNoV的流行基因亚型),分别获得了保守区域。最高相似性的保守区域位于GI族和GII族HuNoV的-连接处,可用于后续引物和探针的设计(数据未展示)。
《3.2. GI 和 GII 族保守区的核苷酸序列》
3.2. GI 和 GII 族保守区的核苷酸序列
为了设计检测HuNoV流行株的引物和探针,将每个族的基因组序列进行多重比对,以确定基因族中的最佳保守片段。采用以KF306212.1中5288~5427的核苷酸位置为标准,分析GI族HuNoV的保守区域碱基分布;以KU870455.1中4959~5108核苷酸位置为标准,分析GII族HuNoV的保守区域碱基分布,统计90条GI族HuNoV序列和132条GII族HuNoV序列上述片段中每个碱基的出现频率,每个位置的A、T、C、G计数如图1和图2所示。本研究设计的引物和探针(橙色)与Kageyama等[13]报道的引物和探针(灰色)进行了比较。NoV基因组中原GI族引物/探针所在位置和碱基已经发生变化,导致其扩增效率始终不高。基于现有流行病毒株序列比对结果,结合Tm 值、G+C含量(%)、扩增效率和基因组间的交叉反应等因素,我们设计了GI和GII族HuNoV新的检测引物和探针(表1)。
《图1》

图1. GI族保守区域每个位置的核苷酸分布图。
《图2》
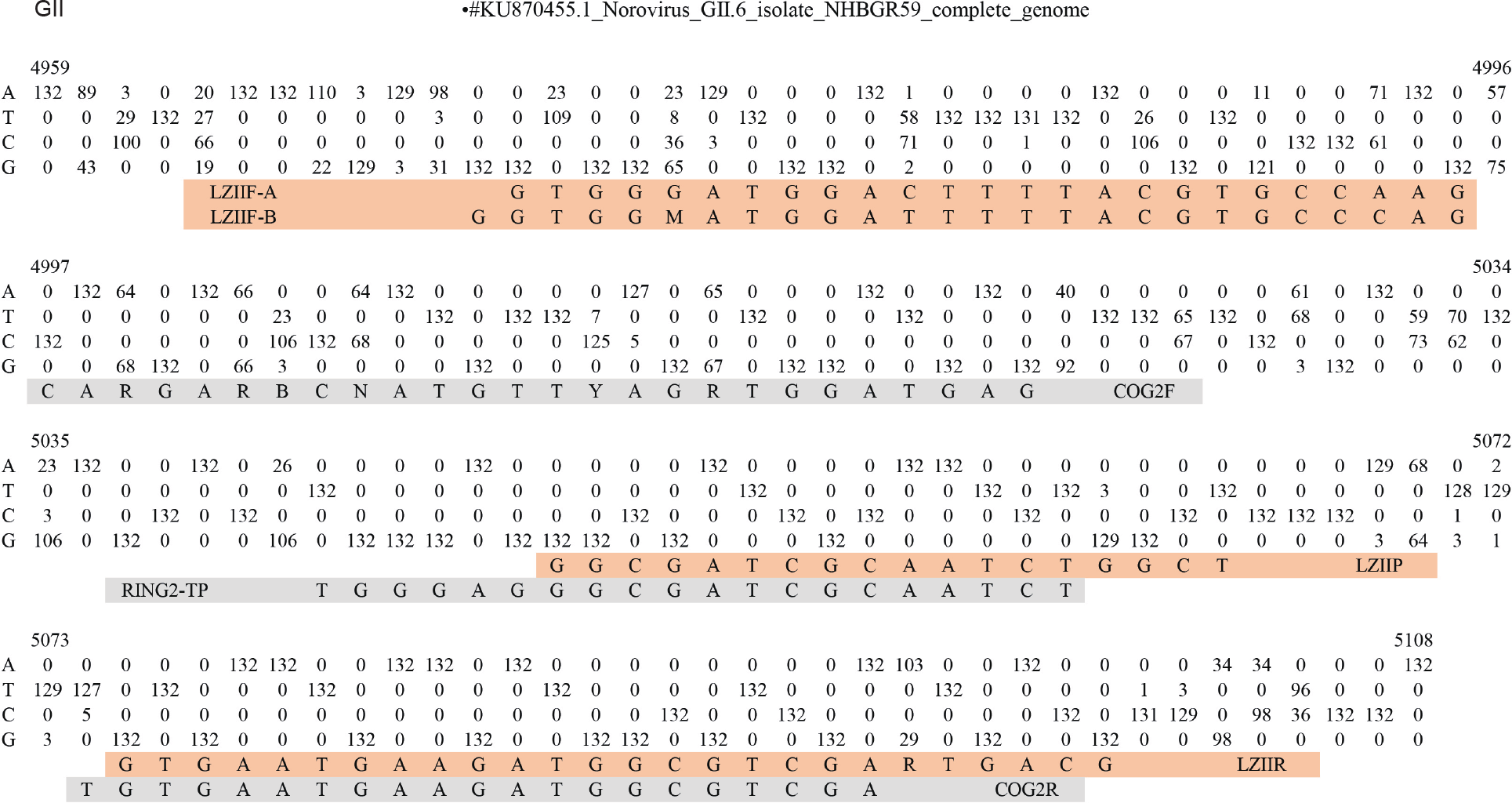
图2. GII族保守区域每个位置的核苷酸分布图。
《3.3. HuNoV 的荧光定量标准曲线》
3.3. HuNoV 的荧光定量标准曲线
以HEX和FAM分 别 标 记GI族(LZIP) 和GII族(LZIIP)探针,实现在一个反应管内同时检测两种基因族HuNoV。用新制备的GI和GII族体外转录的RNA模板,计算拷贝数后等比例稀释;经RT-qPCR检测,分析数据绘制标准曲线、计算反应相关参数,如图3和图4所示。在检测GI和GII族的HuNoV的样本时,双重检测不存在交叉信号。GI族的引物探针的定量标准曲线为y = −3.569lgx +39.161,R 2 = 1.000,E = 90.6%,GII族FAM信号无响应;GII族的引物探针的定量标准曲线为y= −3.387lgx +38.068,R 2 = 1.000,E = 97.3%,GI族HEX信号无响应。
《图3》

图3. GI族HuNoV检测的荧光定量标准曲线。RFU (relative fluorescence unit):相对荧光单位。
《图4》

图4. GII族HuNoVs检测的荧光定量标准曲线
《3.4. 三种 RT-qPCR 方法检测 HuNoV 临床样本的比较》
3.4. 三种 RT-qPCR 方法检测 HuNoV 临床样本的比较
本研究比较了检测临床腹泻样品中GI和GII族HuNoV的三种RT-qPCR体系(表2)。在23份HuNoV临床标本中,5例为GI族HuNoV,18例为GII族HuNoV。对于GI族HuNoV,A方法检出率为100% (5/5),B方法无检出(0/5),C方法检出率为40% (2/5)。临床样本GI.2、GI.4、GI.5使用Kageyama的RT-qPCR方法检测呈假阴性结果。针对C方法检测出的两份GI阳性样品,A方法Cq值显著低于C方法(p<0.05),平均ΔCq值为10.26 ± 0.24,表现出更高的灵敏度。对于GII族HuNoV,A方法检出率为100% (18/18),B方法检出率为50% (9/18),C方法检出率为94% (17/18)。样本17151101 (GII.2)在B、C方法中均未检出,且A方法低于C方法的平均Cq值,ΔCq值为2.20 ± 0.51。在B方法检出样本中,A方法的Cq值低于B方法的,平均ΔCq值为6.26 ± 0.64。
《表2》
表2 三种RT-qPCR方法检测HuNoV临床标本的比较结果
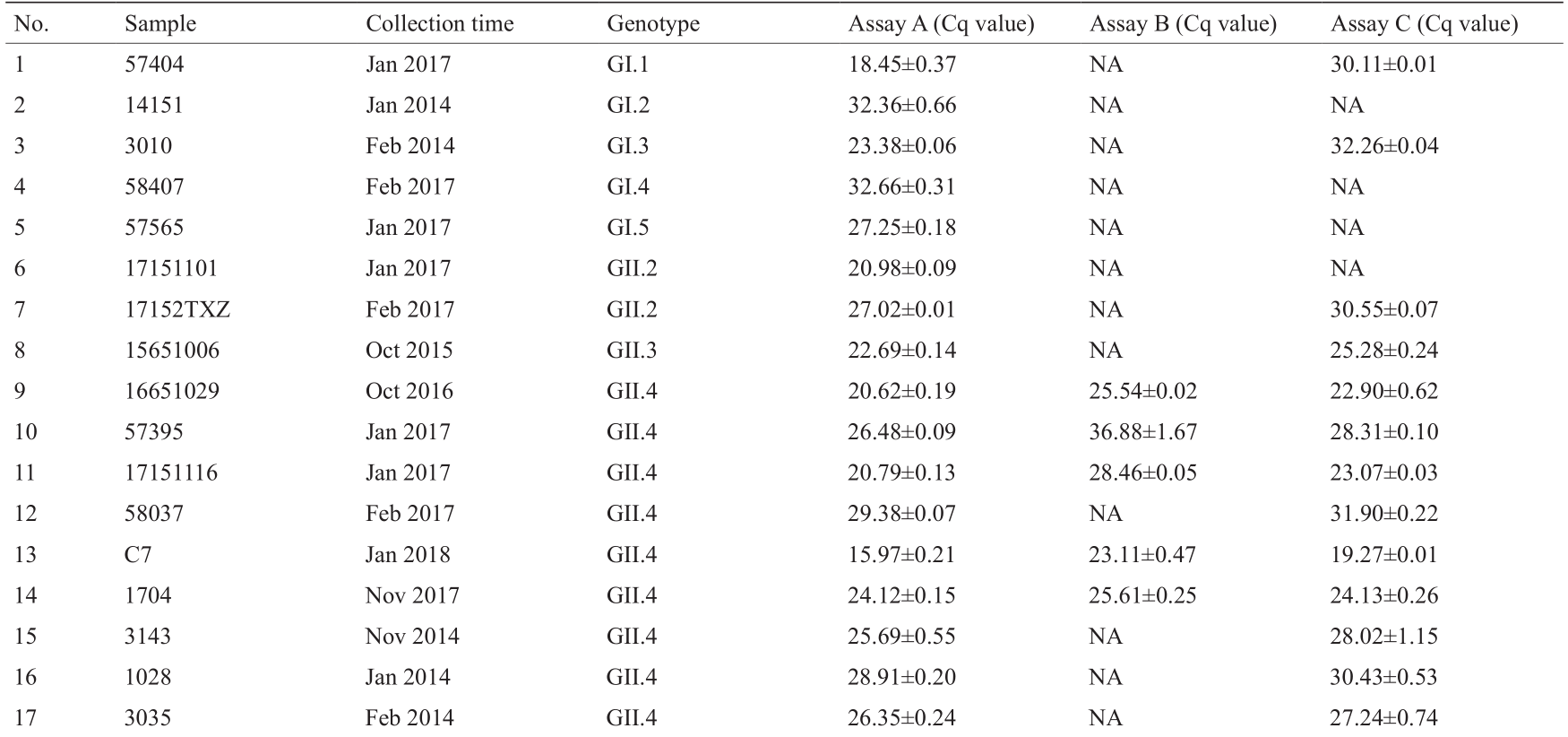

Assay A: ND-RT-qPCR; assay B: primers and probes were replaced with those designed by Kageyama, while the thermal cycle conditions remained the same as for assay A; assay C: Kageyama RT-qPCR. NA: not applicable.
本研究中,A方法可检测所有的临床样本中HuNoV。其他两种检测体系未检出GI.2、GI.4和GI.5型HuNoV。方法B可以检测到很少的GII族HuNoV基因型,对所有GI族HuNoV均未检出。所以,方法A可以检测到HuNoV的基因型,其检出概率(100.0%)高于方法B(39.1%)和方法C(82.6%)。
《4. 讨论》
4. 讨论
HuNoV是导致世界范围所有年龄段的非细菌性腹泻疾病主要病原之一。自2016年以来,只有个别基因型HuNoV可以在肠上皮干细胞内成功复制。因此,暂无有效的HuNoV体外培养体系,极大地阻碍了该病毒的研究进展。目前,HuNoV检测主要依靠ELISA、RT-PCR和RT-qPCR [25]。研究发现:这些方法的灵敏度分别为ELISA 17%、RT-PCR 86%和RT-qPCR 100% [26]。由于分子方法的检测灵敏度较高,常用于HuNoV的检测。
一步法RT-qPCR可在单个PCR管内进行RNA逆转录和cDNA的扩增。另外,RT-qPCR比RT-PCR更为灵敏,并且避免了PCR产物电泳的步骤[25]。许多研究者已经开发了针对GI和GII族HuNoV的RT-qPCR体系,并进行推广使用[13,16–19]。然而,所使用的引物和探针均是基于10~15年前病毒序列设计的。此外,包括保守区域在内的病毒RNA基因组容易发生突变,导致使用的引物/探针与目前的病毒基因组无法结合或结合不良的情况,从而出现假阴性的结果,或由于扩增效率不佳,远低估了病毒滴度。最近,Amarasiri等重新设计了RT-qPCR的检测引物和探针,用于定量分析4种流行病学上重要的基因型GII.3、GII.4、GII.6和GII.17型HuNoV[27]。但是,未提及GII族其他的基因型和整个GI族。本研究中,我们发现传统RT-qPCR方法漏检3份GI族HuNoV样本(表2)。因此,重新设计GI族HuNoV特异性引物/探针显得尤为重要。与临床样本中含有高滴度GII族HuNoV不同[28],GI族HuNoV常见于食品和环境样本[29,30],需要高灵敏度的检测以避免漏检。
为了设计新的引物和探针,我们考虑了如下几个因素:
(1)选定区域片段的保守性,用于设计引物和探针的序列应尽可能保守,尤其是引物3′端的保守性影响后续的PCR延伸;
(2)引物和探针的Tm 值,为了缩短检测周期(将退火和延伸合为一步),根据酶最佳效率在60 ℃,将引物最佳Tm 值调整为60~63 ℃,探针Tm 值比引物高5~10 ℃;
(3)反向引物和探针之间的距离越短,反应过程中的探针酶解效率越高;
(4)为了提高探针杂交的稳定性,本研究选取了鸟嘌呤多于胞嘧啶的反向互补序列用于探针合成;
(5)为了避免GI和GII族病毒之间的交叉反应,本研究重新设计了引物/探针集,以便在一个反应体系中同时检测GI和GII不同族的HuNoV。
我们发现由于引物COG2F(由Kageyama的GII族HuNoV上游引物)可以结合GI族的模板并进行扩增(数据未显示),因此,虽然该区域仍具有较好的保守性(图3、图4),本研究并未根据参考COG2F序列设计新的GII族上游引物。我们发现,GI族保守区域中可完美匹配的引物/探针序列较短,并且Tm 值相对较低。为了实现ND-RT-qPCR在同一反应体系中实现同时检测GI和GII族HuNoV,我们采用TaqMan-MGB作为探针的猝灭基团,3′端的MGB组分增加了探针的Tm 值并可以提高探针的灵敏度和可靠性。5′荧光基团和3′非光化学荧光猝灭基团(non-photochemical fluorescence quenching, NFQ)组合时,NFQ具有较低背景信号的优势,从而可以提高定量的精度。
扩增效率是评估引物/探针组的一个重要指标。对于RT-qPCR反应,扩增效率在90%~110%为可接受范围[31,32]。本研究中,针对GI和GII族HuNoV新引物/探针组的扩增效率分别为90.6%和97.3%。GI族引物探针效率相对较低的原因可能有3个:①GI族完整的基因组数据较少,影响后续的引物探针设计;②HEX探针荧光基团修饰困难,荧光检测时发光的效率较低,后续合成建议换为VIC;③引物和探针的浓度需要进一步优化。目前的效率处于可接受范围内,可以使用。
前期研究表明,RT-qPCR接近检测极限的响应信号,很多是由于PCR制品和(或)PCR污染导致的假阳性结果,并非源自目标片段的扩增[33–35]。在某些情况下,RT-qPCR逆转录和扩增步骤中引物退火温度的降低可能会导致非特异性扩增,从而使探针的背景信号升高。为此,我们通过缩短热循环过程中每个步骤的反应时间,以期减少非特异性探针杂交的概率,并将整体检测周期缩短至约40 min。MIQE指南(The MIQE guidelines: Minimum Information for Publication of Quantitative Real Time PCR Experiments)认为Cq > 40是“阳性但不可量化”[23]。因此,本研究将40个循环设为临界值。
本研究以自主设计的HuNoV RNA模板进行ND-RT-qPCR体系的灵敏度评估。与以前报道的定量标准曲线绘制不同[17,36,37],本研究以两条大于1500 nt体外转录的RNA作为模板,代替先前的质粒模板。与短的DNA和质粒相比,体外转录产生的长片段RNA可以更好地还原病毒RNA的二级结构;同时,生成的标准曲线还可以进一步反映一步法RT-qPCR检测方法逆转录部分中病毒RNA逆转录的效率。使用长片段病毒RNA作为模板,从100 到106 拷贝生成标准曲线,R2 =1.00;当 测 试100 拷 贝GI和GII族 时,Cq值 分 别 为39.10和37.90,进一步稀释病毒RNA模板后未检测到信号(图3、图4)。结果表明,ND-RT-qPCR的灵敏度为100 拷贝,比Kageyama等RT-qPCR的灵敏度高10倍。
以HuNoV临床样本对引物探针进行验证时,我们还发现ND-RT-qPCR相较传统方法展现出更好的性能。ND-RT-qPCR对GI和GII族HuNoV的检出率为100.0%。但是,Kageyama的RT-qPCR方法只在5个GI族HuNoV样本中检测到两个样本;意味着传统的引物/探针组已经不适用于检测目前的GI族病毒样本,且检测灵敏也较低。在之前的文献中也有类似的报道[18,38,39]。与传统的RT-qPCR方法相比,在每个测试样品中,较低Cq值反映出ND-RT-qPCR方法的灵敏度和扩增效率较高。
本研究参照一步法RT-qPCR试剂盒的建议,将引物的Tm 值设为60~63 ℃;正如一步法RT-qPCR试剂盒的建议,在60 ℃温度下完成退火和延伸步骤,避免耗时的反复升降温过程。因此,ND-RT-qPCR可以在40 min内完成。为了评估Kageyama的引物/探针组能否适应快速热循环反应,我们进行了方法B的试验。结果表明,GI族HuNoV样品检出率为0 (0/5),GII族HuNoV样本漏检率为50% (9/18);显示出Kageyama的引物/探针不适合缩短检测时间。
综上所述,本研究建立的ND-RT-qPCR体系可用于临床样本中HuNoV的高效检测,并可以进一步用于环境和食品样品(尤其是新鲜产品或贝类中)中HuNoV的检测与监测。我们的实验数据表明,本研究所设计的ND-RT-qPCR体系具有特异性高、灵敏度高且耗时短(40 min)等特点,可以适用于目前的GI和GII族HuNoV的快速检测。
《致谢》
致谢
本工作得到了中国科学技术部(2017YFC1601200)、国家自然科学基金(31772078)和上海交通大学Agri-X交叉学科基金(2017)的共同支持。
Compliance with ethics guidelines
Danlei Liu, Zilei Zhang, Qingping Wu, Peng Tian, Haoran Geng, Ting Xu, and Dapeng Wang declare that they have no conflicts of interest or financial conflicts to disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




