
2022年1月28日,DeepMind公司宣布向其AlphaFold蛋白质结构数据库(AlphaFold数据库)中加入27种生物的蛋白质组。AlphaFold数据库是一个面向科学家的免费在线数据库。总部位于伦敦的DeepMind是谷歌母公司Alphabet旗下专攻人工智能(AI)的子公司。DeepMind公司根据世界卫生组织的要求,重点选择了这些蛋白质组。换言之,他们公布了导致如麻风病、血吸虫病等罕见热带疾病的生物蛋白质组,以及其他极具耐药性疾病的生物蛋白质组。宣布添加生物蛋白质组的同时,DeepMind团队还说:“我们希望这项举措可以加快研究进程,并支持那些为了消灭此类疾病而不倦工作的研究人员。”[1]
自2021年7月上线以来,AlphaFold数据库新添加了大量蛋白质组数据[2]。DeepMind公司一开始为21个模式生物提供蛋白质结构预测(图1)[3],包括人类,老鼠,果蝇,玉米、亚洲稻种、大豆及酵母等重要作物,大肠杆菌与白色念珠菌等病原体,以及导致疾病的寄生虫,如克氏锥虫(Trypanosoma cruzi;美洲锥虫病)、婴儿利什曼虫(Leishmania infantum;利什曼病)。此后,AlphaFold数据库中的数据不断得到更新和扩展。
《图1》
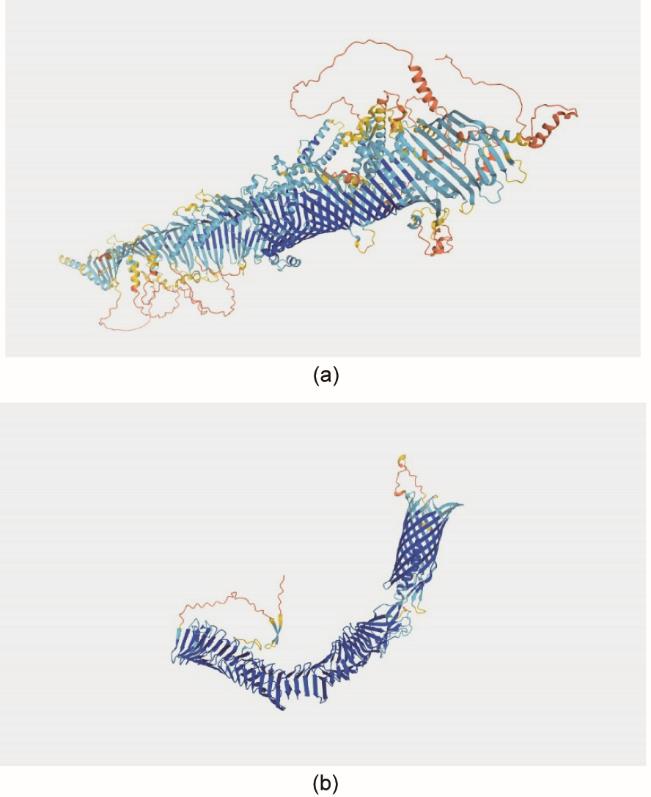
图1 这些由AlphaFold团队生成的图表展示了黑腹果蝇Q9VZS7蛋白(a)以及大肠杆菌P39180蛋白(b)的结构。果蝇和细菌都作为模式生物被广泛用于基础研究。图示为21种模式生物的蛋白质组,这些生物的预测蛋白质组结构被AlphaFold数据库收录。不同的颜色代表AlphaFold团队对构成蛋白质的氨基酸位置预测的置信度,从深蓝色(高置信度)到浅蓝色(中等)、黄色(低),以及橘色(极低)。来源:DeepMind/AlphaFold(公共来源)。
这些进步都表明机器学习为结构生物学领域带来了变革,能够帮助科学家以前所未有的精准度、仅靠蛋白质的基因序列,就能预测蛋白质的形状。如何利用蛋白质基因序列预测蛋白质结构是生物学领域50年来的一个巨大挑战[4‒5]。这项技术具有重要意义,因为用于揭示蛋白质功能的是蛋白质的形状结构,而非其基因序列。准确的蛋白质结构预测为设计高度靶向药物分子,以及培养耐气候变化的作物开启了多种可能。
直到2020年末,DeepMind公司的AlphaFold系统展现了惊人的准确度[4‒5],赢得了两年一次的国际试验——蛋白质结构预测关键评估(CASP)比赛。在这项试验中,多个团队在蛋白质结构预测上一争高下。大部分情况下,预测的蛋白质结构难以与试验确定的结构相区分[4‒5]。
当时没人知道AlphaFold团队会展示多少内容。直到2021年7月,DeepMind公司在《自然》杂志上发表了两篇里程碑式的文章。7月15日,DeepMind公司发表的第一篇文章详细描绘了AlphaFold团队如何克服蛋白质结构在进化、物理和几何上的限制,研发出全新神经网络架构和训练程序,并由此实现结构预测精准度的跨越式提升[6]。在这篇文章发表的同时,DeepMind公司开源发布了AlphaFold代码[2],供全世界科学家使用。
7月22日,DeepMind公司发表了第二篇文章,称其已经对整个人类蛋白质组中98.5%的蛋白质进行了结构预测。相较之下,通过试验确定的人类蛋白质序列不到整个人类蛋白质组的1/5 [7]。同一天,DeepMind公司携手欧洲分子生物学实验室(EMBL)旗下欧洲生物信息研究所(EBI)共同宣布AlphaFold数据库上线[8]。数据库一开始就包含上文中提及的21种模式生物蛋白质组中的36万多种蛋白质预测结构。
随后,2021年12月,DeepMind公司与EMBL-EBI宣布对AlphaFold数据库进行扩展,将UniProtKB/Swiss-Prot数据库中的蛋白质序列导入其中。相比AlphaFold数据库,UniProtKB/Swiss-Prot高质量数据库的内容均为人工标注的科学文献记录[9]。此时,AlphaFold数据库中预测的蛋白质结构数量已经超过80万个。计划2022年进一步更新AlphaFold数据库中蛋白质预测结构,预计预测的蛋白质结构数量将超过一亿个[3]。
这些成就对结构生物学领域“意义非凡”,CASP创始人兼组织者、美国马里兰大学生物科学与生物技术研究所教授John Moult如此评价:“我从未见过哪一个软件的普及速度会如此之快。并不夸张地说,现在所有结构生物学家不是在用AlphaFold数据库,就是在用自己类似的软件。”
美国伊利诺伊州芝加哥大学丰田技术研究所计算生物学教授Jinbo Xu称,AlphaFold数据库对结构生物学家来说非常方便。Xu教授还说:“然而,AlphaFold软件工具本身更重要。” Xu教授研发的蛋白质结构预测软件RaptorX赢得了上一届CASP比赛[10]。
英国牛津大学首席研究员Richard Wheeler表示赞同。Wheeler的实验室探索了真核生物Discoba亚界的单细胞生物利什曼虫和锥虫属寄生虫的基础细胞生物学。他说:“长时间以来,我一直期待能有像AlphaFold数据库这样的产品出现。因为我们与被忽视的热带病原体打交道,但并没有像人类或酵母等模式生物这样通过试验测定数据的数据库,所以我非常兴奋。”
然而,Wheeler立即关注到,基因数据的稀少和对如克氏锥虫(图2)这样研究甚少的生物体了解的缺失会让AlphaFold数据库面临难题。他说:“AlphaFold数据库并不太适合预测Discoba亚界生物体的结构。”通过全面收集Discoba亚界生物体最新、可用的蛋白质序列数据,并将其输入AlphaFold程序,Wheeler在AlphaFold数据库中大幅提高了结构预测效果,然后将成果免费分享给寄生虫学社区[11]。
《图2》
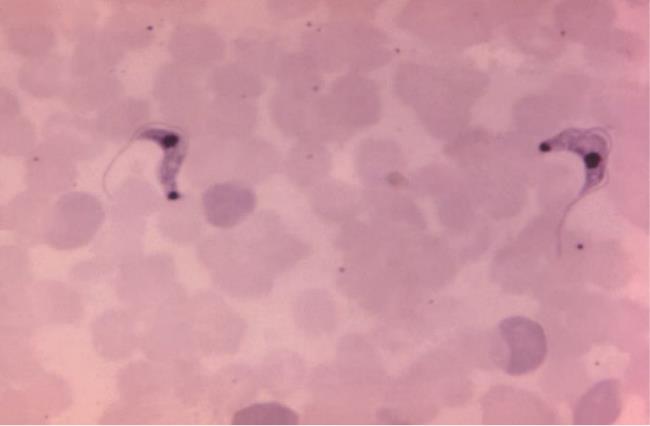
图2 该血涂片显微照片展现了引起美洲锥虫病的单细胞鞭毛寄生虫克氏锥虫。DeepMind公司已经预测了包括克氏锥虫在内的造成热带疾病的病原体的蛋白质组中的蛋白质结构,加快了高效医疗进程。来源:CDC/Myron G. Schultz(公共来源)。
Wheeler认为,该工作对其他使用AlphaFold数据库研究被忽视的生物体结构的生物学家影响极大。他说道:“虽然AlphaFold数据库无法完善近1/3的Discoba亚界生物体蛋白质结构的预测,但是我发现AlphaFold数据库能显著改善剩下2/3的预测结构,并实现了Discoba亚界首例高置信度的生物体结构预测。AlphaFold数据库预测效果非常惊人,影响深远。”
不过,AlphaFold数据库并非近期唯一的开源预测工具。在AlphaFold数据库上线后不久,美国华盛顿大学蛋白质设计研究所(Institute for Protein Design at the University of Washington)生物化学教授David Baker与其同事一起开发并上线了RoseTTaFold数据库。RoseTTaFold数据库在预测准确性方面与AlphaFold数据库相近,但RoseTTaFold数据库消耗的计算机算力更少,因此速度也更快[12]。
此类工具不仅改变了蛋白质结构预测技术,也在相对冷门的核糖核酸(RNA)结构预测领域取得了进展。RNA是一种类似脱氧核糖核酸(DNA)的核酸,仅为单链结构,具有不同功能。RNA在细胞生理学上扮演重要角色。不同种类的RNA执行着无数的生物任务,比如,信使RNA将DNA信息翻译为蛋白质(图3)。
《图3》

图3 DNA中的蛋白质编码基因转录给mRNA,然后被翻译成功能性蛋白,而RNA编码基因直接转录为非编码RNA(ncRNA)。掌握RNA的折叠结构,就像了解蛋白质结构一样,对于理解分子功能如何影响人类健康与疾病具有重要意义。来源:Thomas Shafee(CC BY 4.0)。
对机器学习而言,在仅靠RNA基因序列预测RNA单链的折叠结构上面临的问题与预测蛋白质折叠结构类似,但用于训练机器学习模型的试验确定的RNA结构非常少;科学上可用的已确定的RNA结构不到蛋白质结构的1% [14]。不过,美国加利福尼亚州斯坦福大学的研究人员报道称,他们去年利用原子旋转等变评分器(Atomic Rotationally Equivariant Scorer, ARES)在此领域取得了重大进展。
斯坦福大学的研究人员利用机器学习方式,采用仅由18个RNA分子的结构配置组成的数据训练了ARES。与AlphaFold团队的蛋白质训练方式不同,ARES的训练并不包括与RNA分子的折叠以及行为模式相关的非特定领域信息,仅使用RNA分子的原子相对坐标。当一个RNA分子的特定基因序列带有未知(对ARES未知)结构时,系统将会使用名为Rosetta FARFAR2 [13]的开源RNA建模工具,生成超过1500个候选RNA分子结构。根据训练结构,系统随后挑选自认为最接近现实的结构。根据未知RNA结构预测挑战赛RNA-Puzzles(类似于CASP比赛)的预测模型基准,ARES相比其他结构预测手段表现得更出色。该团队在2021年8月将他们的成果发表在《科学》杂志上[14]。
论文第一作者Raphael Townshend说道:“在结构生物学中,你可以认为一个原子就是一个机器学习的基础数据类型。”Townshend离开了斯坦福大学,在旧金山成立了Atomic AI生物科技公司并担任该公司的CEO。Atomic AI公司主要利用机器学习手段,设计新分子、新药物。Townshend说道:“我们成功地调整了蛋白质领域的机器学习模型,并将其运用在RNA领域。它的效果十分出色,有力地证明了机器学习的普适性。”
ARES代表现有RNA结构预测系统的进一步提升,但正如美国教堂山北卡罗来纳大学化学系教授Kevin M. Weeks在《科学》的一篇前瞻性文章中所说:“ARES依然不能稳定达到原子级解析度,也不能指导识别关键功能点或药理发现工作。”[15]
原先在DeepMind公司AlphaFold团队工作的Townshend也承认了这一点。他说道:“论精准度,ARES网络世界第一。即使如此,它也只是合理的药物发现之路的第一步。然而,它可以直接作为有力的筛选工具,在试验中使用。”Townshend说,他想在RNA领域实现像蛋白质领域一样的成果——借助AI的力量,在短短数年内大幅提升精准度。然而,在不使用特定领域信息的情况下,尚不明确这些模型能否实现同样等级的精准度。
无论如何,蛋白质预测上的成就,以及开源工具的不断丰富,都为人们解决RNA折叠问题提供了不少帮助。CASP15于2022年5月启动,主题包括RNA分子结构预测。Moult说道:“我们正一如既往地根据新成果不断进行调整,并与RNA-Puzzles团队一同合作,吸引更多人。蛋白质研究人员也将对进入RNA领域感兴趣。”
CASP15也更侧重预测蛋白质复合体结构。Xu说道,这就是该领域前进的方向,因为蛋白质并非独立存在。“蛋白质通过与其他蛋白质和分子相互作用而变得独特,因此我认为这(预测蛋白质复合体结构)比预测单个蛋白质结构更重要。这是在各种行业应用,尤其在药物设计中需要解决的基础性问题。”











 京公网安备 11010502051620号
京公网安备 11010502051620号




