
《1. Introduction》
1. Introduction
Cancer is a global public health problem that causes the death of tens of thousands of people each year. Despite enormous efforts to develop effective treatment strategies, no satisfactory results have yet emerged, and the overall survival rate and disease-free survival of most cancers remain poor. Although conventional treatments, including radiotherapy, chemotherapy, surgical operation, and molecular-targeted therapy, are effective in attacking tumor cells, they simultaneously create life-threatening side effects. The most common adverse effects include gastrointestinal reaction and bone marrow suppression, which not only influence the postoperative quality of life, but can also lead directly to death [1]. In the last decades, immunotherapy has been an active area of research in tumor therapy, and considerable efforts have been expended in this field. These new strategies, which are represented by immune checkpoint inhibitors (e.g., programmed death 1 (PD-1), programmed death ligand 1 (PD-L1), and cytotoxic T lymphocyte-associated antigen 4 (CTLA-4)) and chimeric antigen receptor T cell (CAR-T) immunotherapy, have shown inspiring efficacy in addressing various types of cancer [2–8]. Unfortunately, an increasing number of studies have demonstrated that only 20%–30% of patients respond to these strategies [9], bringing up the question of why immunotherapies have failed in 70%–80% of patients. One explanation that has been accepted is that tumors have evolved an immune-suppressing network to prevent attacks from immune systems. Regulatory T cells (Tregs) are an important part of this network. Tregs that strongly express CD25 (interleukin (IL)-2 receptor α-chain) and forkhead box P3 (FOXP3) are enriched in the tumor microenvironment, and primarily comprise an immunosuppressive network [10]. Here, we focus on the critical roles played by Tregs in tumor immunosuppressing network, and on their therapeutic potential to treat diverse types of tumor.
《2. Treg differentiation and development》
2. Treg differentiation and development
Based on their origins, Tregs can be characterized into two populations. The first class of Tregs are natural regulatory T cells (nTregs), which differentiate from the thymus and are induced by a broad spectrum of autoantigens [11]; for this reason, nTregs are also known as thymus-derived Tregs (tTregs). After being stimulated by the major histocompatibility complex (MHC)– autoantigen complex, a fraction of CD4+ CD8+ T cells in thymocytes start to express CD25, through which IL-2 induces the production of FOXP3 via signal transducer and activator of transcription 5 (STAT5) [12]. The other class of Treg comprises induced regulatory T cells (iTregs), which are also known as peripherally derived Tregs (pTregs). Many factors (e.g., transforming growth factor β (TGF-β), dendritic cells (DCs) expressing indole amine 2,3-dioxygenase (IDO), or retinoic acid) can convert peripheral CD4+ naive T cells into iTregs [13–15]. Both subsets have similar surface markers and considerable suppressive function against effector T cells (Teffs). However, accumulating evidence has shown that there are many differences between Treg subsets, such as different protein expression and epigenetic modification. For example, iTregs reportedly express a low level of Helios and Neuropilin-1 (NRP1), whereas nTregs are abundant in these molecules [16,17]. In addition, there are two other special subsets of Tregs that do not express Foxp3: IL-10-producing T regulatory type 1 (Tr1) cells and TGF-β-producing T helper type 3 (Th3) cells [18].
In the process of Treg development, T cell receptor (TCR) signal is indispensable, such that blockage of TCR leads to suppressed Treg development [19]. T cells with a high affinity to autoantigens undergo negative selection in the thymus, leading to apoptosis, whereas T cells with a low affinity for autoantigens survive and develop into Teffs. The threshold of avidity that is selected to produce nTregs is between positive and negative selection [20]. In addition, Treg development requires co-stimulatory molecules such as CD28 and glucocorticoid-induced tumor necrosis factor receptor (TNFR)-related protein (GITR). Previous studies have revealed that in mice lacking CD28 or CD80/CD86, the Treg number is decreased [21,22]. Another indispensable factor for Treg development is IL-2. Tregs remain non-responsive to high-dose IL-2, solid-coated or soluble anti-CD3 monoclonal antibodies (mAb), or a combination of anti-CD3 mAb and anti-CD28 mAb. Only when a TCR signal and a high concentration of exogenous IL-2 are simultaneously present can a Treg be activated; however, the proliferation is weaker than that of CD4+ CD25– T cells [23]. In fact, a negative loop is present in which antigen-activated T cells produce IL-2 to induce the proliferation of Tregs, which then mount a suppressory response against these activated T cells in order to avoid overreaction. The nuclear factor-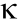 B (NF-B) signaling pathway, downstream of TCR/CD28, also plays a critical role in Treg development. Under conditions of TCR/CD28 co-stimulation, C-REL binds to the promoter and conserves non-coding DNA sequences of the Foxp3 gene in order to regulate Foxp3 transcription [24]. Medullary thymic epithelial cell (mTEC) signaling is also an important factor in regulating the development of nTregs. A deficiency in TNFR associated factor 6 (TRAF6) impairs the maturation of mTECs, and thereby inhibits Treg development [25]. In a mouse model of cancer, researchers found that NRP1 plays a vital role in maintaining the function of tumor-infiltrating Tregs. Selective knockout of Nrp1 in mice results in a loss of function of Tregs. The mechanism underlying this phenotype is that the interferon-γ (IFN-γ) produced due to a deficiency in Nrp1 drives the fragility of tumorinfiltrating Tregs [26]. In this regard, it is notable that a lack of Nrp1 simply influences the antitumor function of Treg; its role in avoiding autoimmunity disease is not affected.
B (NF-B) signaling pathway, downstream of TCR/CD28, also plays a critical role in Treg development. Under conditions of TCR/CD28 co-stimulation, C-REL binds to the promoter and conserves non-coding DNA sequences of the Foxp3 gene in order to regulate Foxp3 transcription [24]. Medullary thymic epithelial cell (mTEC) signaling is also an important factor in regulating the development of nTregs. A deficiency in TNFR associated factor 6 (TRAF6) impairs the maturation of mTECs, and thereby inhibits Treg development [25]. In a mouse model of cancer, researchers found that NRP1 plays a vital role in maintaining the function of tumor-infiltrating Tregs. Selective knockout of Nrp1 in mice results in a loss of function of Tregs. The mechanism underlying this phenotype is that the interferon-γ (IFN-γ) produced due to a deficiency in Nrp1 drives the fragility of tumorinfiltrating Tregs [26]. In this regard, it is notable that a lack of Nrp1 simply influences the antitumor function of Treg; its role in avoiding autoimmunity disease is not affected.
《3. Treg function regulation》
3. Treg function regulation
FOXP3 is a master regulator in the development and function of Tregs [27]. The importance of FOXP3 is highlighted by the profound loss of nTregs in Foxp3-knockout mice. In addition, the adoptive transfer of Foxp3 can convert peripheral CD4+ CD25– T cells into iTregs. Both of these findings indicate that Foxp3 is a sine qua non in Treg development. Foxp3 expression is regulated by a series of physiological signals and protein modifications. However, in addition to Tregs, there are multiple cell types that express Foxp3 but lack a suppressive function, including epithelial cells of the breast, lung, and prostate [28]. This demonstrates that the expression of Foxp3 is insufficient for establishing the Treg cell lineage. Furthermore, in some cases, FOXP3– cells also possess a potent inhibitory function [29]. An increasing amount of evidence has revealed that there are two independent events in the process of Treg development: the expression of Foxp3 and the modification of Foxp3 [30].
《3.1. Regulation of Foxp3 at the transcriptional level》
3.1. Regulation of Foxp3 at the transcriptional level
Foxp3 gene expression is controlled by four elements: the promoter region; two conserved non-coding sequences (CNS1 and CNS2) within the first intron; and the second intron, which contains CNS3 [31]. CNS1 contains binding sites for activator protein 1(AP-1), mothers against decapentaplegic homolog 3 (SMAD3), and the nuclear factor of activated T cells (NFATs). In CNS1- knockout mice, the expression of Foxp3 in tTregs is comparable with that in wild-type (WT) mice, while iTreg is reduced; this finding reveals the role of CNS1 in the peripheral induction of Tregs. CsA (an inhibitor of NFAT) and SIS3 (an inhibitor of SMAD3) can result in a loss of Foxp3 expression [32]. CNS2, which contains binding sites for STAT5 [33], cyclic adenosine monophosphate (cAMP) responsive element-binding (CREB)/activating transcription factor (ATF) [34], forkhead box protein O1 (FOXO1), and forkhead box protein O3 (FOXO3) [35], is termed as ‘‘Treg-specific demethylated region” (TSDR), because this area in nTregs is completely demethylated. CNS2-deficient mice demonstrate a decreased number of Tregs after six months, suggesting that CNS2 is essential in the maintenance of stability but dispensable in the expression of Foxp3 [32]. CNS3 is bound by C-REL, which acts as a chromatin opener to promote the formation of a Foxp3-specific enhanceosome; the number of Tregs is reduced in  mice [24].
mice [24].
《3.2. Post-modification of Foxp3 in Tregs》
3.2. Post-modification of Foxp3 in Tregs
Post-translational modification of Foxp3 in Tregs is comprised of ubiquitination, acetylation, methylation, poly(ADP-ribosyl)ation, and so forth. Under inflammation elicited by pro-inflammatory cytokines and lipopolysaccharide, Tregs will downregulate the expression of Foxp3, thus permitting the gain of Teff-like functions [36]. The mechanism underlying this phenotype is as follows: During inflammation, the E3 ubiquitin ligase Stub1 is activated and induces the degradation of FOXP3 in an Hsp70-dependent manner. In contrast, ubiquitin-specific protease 7 (USP7) and ubiquitinspecific protease 21 (USP21) are two major de-ubiquitin enzymes that are capable of reducing the number and function of Tregs [37,38].
The balance between lysine acetylation and deacetylation is another factor that plays an essential role in regulating the stability and function of FOXP3 proteins. The histone acetyltransferases (HATs) TIP60 [39] and p300 [40] can stabilize the FOXP3 protein, thus promoting its inhibitory function, via FOXP3 acetylation. Here, nicotinamide adenine dinucleotide (NAD)-dependent deacetylase sirtuin-1 (SIRT1) has an opposite role to those of TIP60 and p300 [41]. Compared with conventional T cells, CNS2 and Foxp3 promoter contain a highly demethylated region that is indispensable in maintaining the stability of Foxp3 expression [42]. Methylation of the CREB/ATF site in the first intron shows a negative correlation with Foxp3 expression. There are many other adjustment methods as well, such as phosphorylation and small ubiquitin-like modifier (SUMO) modification. This is a complicated process regulated by multiple elements.
《4. Treg functions and suppressive mechanisms》
4. Treg functions and suppressive mechanisms
《4.1. Suppressive mechanisms of Tregs》
4.1. Suppressive mechanisms of Tregs
There are diverse mechanisms by which Tregs exert their suppressor activity (Fig. 1). Among these, some proposed mechanisms are listed below.
《Fig. 1》

Fig. 1. Suppressive mechanisms of Tregs. GITRL: GITR ligand; LAG3: lymphocyte activation gene 3 protein; GZ-B: granzyme B; TIGIT: T cell immunoglobulin (Ig) and immunoreceptor tyrosine-based inhibition motif (ITIM) domain.
4.1.1. Secretion of Tregs
Tregs can secret various inhibitory cytokines such as IL-10, TGF-β [43], and IL-35 [44]. At present, the view that IL-10 and TGF-β may participate in the regulatory activity of Tregs is still controversial. IL-10 and TGF-β can relieve intestinal inflammation caused by Treg deficiency [45]. However, neutralization of IL-10 and TGF-β in vitro does not impair the inhibitory effect of Tregs [46]. Collison et al. [44] have demonstrated that the co-culture of Tregs and Teffs leads to upregulation of IL-35, consisting of Epstein-Barr virus induced gene 3 (EBI3) and IL-12A, and that the ectopic expression of IL-35 can suppress the functions of Teffs. It has now been well substantiated that IL-35 contributes to abrogation of inflammatory bowel disease and psoriasis [47]. Neutralization of IL-35 can exert a limitation on tumor growth in multiple murine models of human cancer [48]. Furthermore, Treg can be immune suppressive by secreting granzyme B (GZ-B). GZ-B is a serine protease with the ability to induce apoptosis in targeted cells [49]. Compared with WT mice, the Tregs derived from 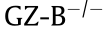 mice have a lower suppressive ability [50].
mice have a lower suppressive ability [50].
4.1.2. Direct contact between cells
Tregs can also function through direct contact between cells via TGF-β, CTLA-4, GITR, and galectin-1. CTLA-4 is a co-inhibitory molecule expressed by both Tregs and Teffs. Activation of Teffs requires co-stimulatory signals supplied by a combination of CD28 and CD80/CD86. Affinity of CTLA-4 is higher than that of CD28, thus restraining combination between CD28 and CD80/CD86 [51–53]. GITR is a co-stimulatory molecule that is constitutively overexpressed in CD4+ CD25+ Tregs. GITR-knockout mice show an impaired Treg function [54]. Galectin-1, which belongs to the family of β-galactoside-binding proteins, is a negative growth factor that regulates cell proliferation. Galectin-1 secreted on the surface of Tregs interacts with Teffs, destroying their cell cycle progress and thus inducing their apoptosis. Blockade of galectin-1 reduces the efficacy of Tregs in both human and mice [55].
4.1.3. Extracellular adenosine triphosphate
The extracellular adenosine triphosphate (ATP) produced by damaged cells acts as a ‘‘natural adjuvant” in inflammation reactions. Extracellular enzymes CD39 and CD73 can degrade ATP to adenosine monophosphate (AMP) and produce adenosine (an inhibitor of Teff activation) [56,57].
4.1.4. Competition between Tregs and conventional T cells
IL-2 is a growth factor required by both Tregs and Teffs. Tregs cannot produce IL-2 itself because of the expression of key transcription factor FOXP3, which is a transcriptional repressor of the IL-2 gene. Therefore, competition between Tregs and conventional T cells is one of the mechanisms underlying Treg suppressive efficacy [58].
4.1.5. Dendritic cells
DCs are antigen-presenting cells responsible for the presentation of antigens to T cells. Tregs can interact with DCs via lymphocyte activation gene 3 protein (LAG3) [59], T-cell immunoglobulin (Ig) and immunoreceptor tyrosine-based inhibition motif (ITIM) domain (TIGIT) [60], and NRP1 [61] to induce the production of immunosuppressive IDO [62]. IDO degrades tryptophan and lead to apoptosis in T cells.
4.1.6. Oxidative stress
Tregs in tumors undergo death under oxidative stress. It is notable that apoptotic Tregs are still immunosuppressive, which presents a challenge to antitumor immunotherapy targeting Tregs. The mechanism may be due to the release of large amounts of small metabolites (e.g, ATP). In general, ATP is helpful to the host; however, Tregs in a sudden death state quickly convert ATP to adenosine, which then binds to receptors on the T cell surface and affects T cell function [63].
《4.2. Treg functions in tumors》
4.2. Treg functions in tumors
The best-known function of Tregs is that of suppressing. That is, after being stimulated by a TCR signal, Tregs possess the capacity to inhibit the activation and proliferation of multiple cells including Teffs, B cells, natural killer (NK) cells, and DCs. Moreover, the immunosuppressive effects of Tregs are not antigen specific and are not restricted by MHC. Numerous studies have demonstrated that Tregs participate in homeostatic regulation and tumor immune escape. The overwhelming evidence from multiple studies in both mice and humans indicates that adoptive Treg transfers can reverse autoimmune disease that is induced by Treg deficiency or Foxp3 mutation [64–66]. Therefore, Tregs are essential and indispensable in protecting the host from dysregulation.
In tumors, it has been recognized that the abundant presence of Tregs often indicates poor prognosis [67]. In order to identify the common basis between tumor immunity and autoimmunity, Shimizu et al. [10] demonstrated that removing Tregs via antiCD25 antibody can effectively alleviate a variety of inoculated syngeneic tumors. However, recent new studies have challenged the relationship between Tregs and tumor prognosis. Researchers have found that based on the expression of Foxp3 and CD45RA, Tregs can be classified into three types: naive Tregs (FOXP3loCD45RA+ ), effector Tregs (FOXP3hiCD45RA– ), and non-Tregs (FOXP3loCD45RA– ). Naive Tregs are only weakly suppressive, while effector Tregs, which differentiate from naive Tregs after antigenic stimulation, possess strong suppressive activity and stable function. Non-Tregs cannot exert an inhibitory effect but can secrete pro-inflammatory cytokines. Based on this classification, colorectal cancer (CRC) has been separated into two types, one of which is predominantly infiltrated by effector Tregs, while the other is abundant in non-Tregs. In the former group, Tregs indicate a poor prognosis; however, in the latter group, patients with more Treg infiltration in the tumor microenvironment have a better prognosis [68,69].
In fact, the appearance of Tregs is not always harmful. Strong evidence supports the notion that chronic inflammatory disease promotes the progression of certain types of cancers such as colon cancer, liver cancer, and pancreatic cancer. Thus, methods targeting the inflammatory network of the tumor environment may be able to offer tumor prevention and treatment [70]. Tregs are one of the main anti-inflammatory factors. Epidemiological data have revealed that individuals exposed to diverse environmental organisms maintain a protective Treg phenotype that prevents cancer, whereas hygienic individuals with little exposure early in life suffer an increased risk of malignancy later in life due to a dysregulated Treg feedback loop [71].
Furthermore, an increasing number of studies have revealed other unique functions of FOXP3+ Tregs. One such function is regulating the adipose-associated inflammation and metabolic process through the peroxisome proliferator-activated receptor-γ (PPAR-γ) and IL-33–suppression of tumorigenicity 2 (ST2) axis [72,73], which may promote tissue repair in non-lymphoid organs via the secreted cytokine amphiregulin (Areg) [74,75] or stimulate the differentiation of stem cells [76]. These findings provide new possibilities that the roles of tumor-associated Tregs may be diverse; new studies focusing on these additional functions may reveal therapeutic targets for antitumor clinical studies.
《5. Immune cells and the tumor microenvironment》
5. Immune cells and the tumor microenvironment
One of the prominent features of tumor cells is their unlimited capability for proliferation, which leads to a large energy demand. There are two major pathways for cells to gain ATP. One is the glycolysis (Gly) of glucose in cytosol, and the other is the oxidative phosphorylation of glycosides in mitochondria via the tricarboxylic acid (TCA) cycle. Tumor cells preferentially select the glycolytic pathway to obtain ATP and other forms of energy, even in an aerobic environment, which is known as the Warburg effect [77]. Through the glycolytic pathway, tumor cells produce a large number of metabolites such as lactic acid and carbon dioxide. These products not only inhibit the effect of conventional T cells, but also inhibit the maturation and activation of DCs. The reason is that antitumor immune cells (such as cytolytic T lymphocytes (CTLs), Teffs, and activated DCs), like tumor cells, obtain the raw materials needed for energy and biosynthesis through aerobic Gly and glutamine decomposition pathways [78]. However, Tregs prefer the fatty acid energy supply pathway, and can use the metabolites of neoplasms to obtain energy [79]. Thus, tumor cells proliferate in an immunosuppressive network in which Tregs are abundant, but T cells and DCs lose their function. Chang et al. [80] have found that tumor cell and T cell metabolism of glucose in the tumor microenvironment can inhibit the activation of the mammalian target of rapamycin (mTOR) signaling pathway of T cells and decrease the number of IFN-γ, thereby causing T cells to exhibit low reactivity in the tumor microenvironment. Given these findings, it is suggested that in a tumor environment, Tregs form a symbiotic relationship with tumor cells.
Other than metabolic causes, there are many other triggers of Treg aggregation in the tumor microenvironment. Tumor cells and infiltrating macrophages can secrete a variety of chemokines (e.g., CC chemokine ligand 22 (CCL22)/CC chemokine receptor 4 (CCR4) [81] and CXC motif chemokine 12 (CXCL12)/CXC chemokine receptor 4 (CXCR4) [82]) to recruit Tregs. Furthermore, DCs and some suppressors including IL-10, TGF-β, and IDO, which are enriched in the tumor environment, are able to convert Teffs into CD4+ CD25+ FOXP3+ T cells and amplify thymic-derived Tregs.
《6. Unique features of tumor-associated Tregs》
6. Unique features of tumor-associated Tregs
An improved understanding of the unique features of tumorinfiltrating Tregs is of great importance for therapeutic benefit. Plitas et al. [83] have found that relative to normal breast parenchyma (NBP) and peripheral blood, tumor-resident T cells exhibit an increased abundance and highly proliferative phenotype of FOXP3+ Tregs. Furthermore, more advanced grades and more aggressive forms of breast cancer appeared to contain a heightened presence of activated Tregs with potent suppressor function. RNA-seq analysis of purified CD25hiCD4+ Tregs and CD25– CD4+ Teffs indicated that the gene expression patterns of tumor-resident Tregs and Teffs cells were distinct from those of corresponding cells isolated from peripheral blood. Detailed analysis has identified chemokine (C–C motif) receptor 8 (CCR8) as the most robustly and differentially expressed chemokine receptor in breast tumor-resident Tregs. The correlation of a high Ccr8/Foxp3 mRNA expression ratio with markedly decreased disease-free and overall survival levels was observed as well; together, these findings indicate that CCR8 may serve as a promising target for antitumor immunotherapy. The observed protective antitumor immunity in subsequent research that targeted CCR8 in colon cancer strongly indicated the biological importance of this receptor [84]. Several genes not previously implicated in Tregs biology were also found to be robustly expressed by tumor Tregs, such as the melanoma antigen family H1 (MAGEH1) gene and CD177. In parallel studies, both De Simone et al. [85] and Zheng et al. [86] found a series of genes that were preferentially upregulated by intra-tumoral Tregs. It is notable that 31 of the different genes were identified in all three studies cited above, including CTLA-4, tumor necrosis factor receptor superfamily, member 4 (OX40), GITR, 4-1BB, TIGIT, ICOS, and CD27. However, because these cell surface markers are also expressed in a fraction of non-tumor-infiltrating Tregs, the functional consequences for antitumor immunotherapy against these receptors remain unclear.
《7. Tregs in antitumor immunotherapy》
7. Tregs in antitumor immunotherapy
《7.1. Checkpoint blockades associated with Tregs》
7.1. Checkpoint blockades associated with Tregs
Clinical usage of immune checkpoint inhibitors, such as CTLA-4, PD-1, and PD-L1, can induce robust and durable responses in many cancer types. In tumor patients treated with Ipilimumab, a blockade of CTLA-4, the number of Tregs infiltrating the tumor was reduced [87,88]. Furthermore, the CTLA-4 signal is essential for both Tregs and Teffs, so that administration of therapeutic antiCTLA-4 antibody not only attenuates the immune-suppressive function of Tregs, but also augments Teff response [89]. In addition, the mechanisms of anti-CTLA antibodies have recently been challenged by increased researches that demonstrate that these antibodies may function by inducing antibody-dependent cellular cytotoxicity (ADCC), rather than by blocking inhibitory checkpoints. It is noteworthy that in many types of cancers, such as CRC, immune-suppressive Tregs may play a critical role in restricting cancer progression; therefore, the depletion of tumorinfiltrating Tregs in these cancers could be unadvisable. The role of anti-PD-1 antibody in blocking Tregs has not yet been determined. In a mouse model, it has been reported that the suppressive function of PD-1-expressing Tregs is higher than that of Tregs without PD-1 expression [90] (Fig. 2).
《Fig. 2》
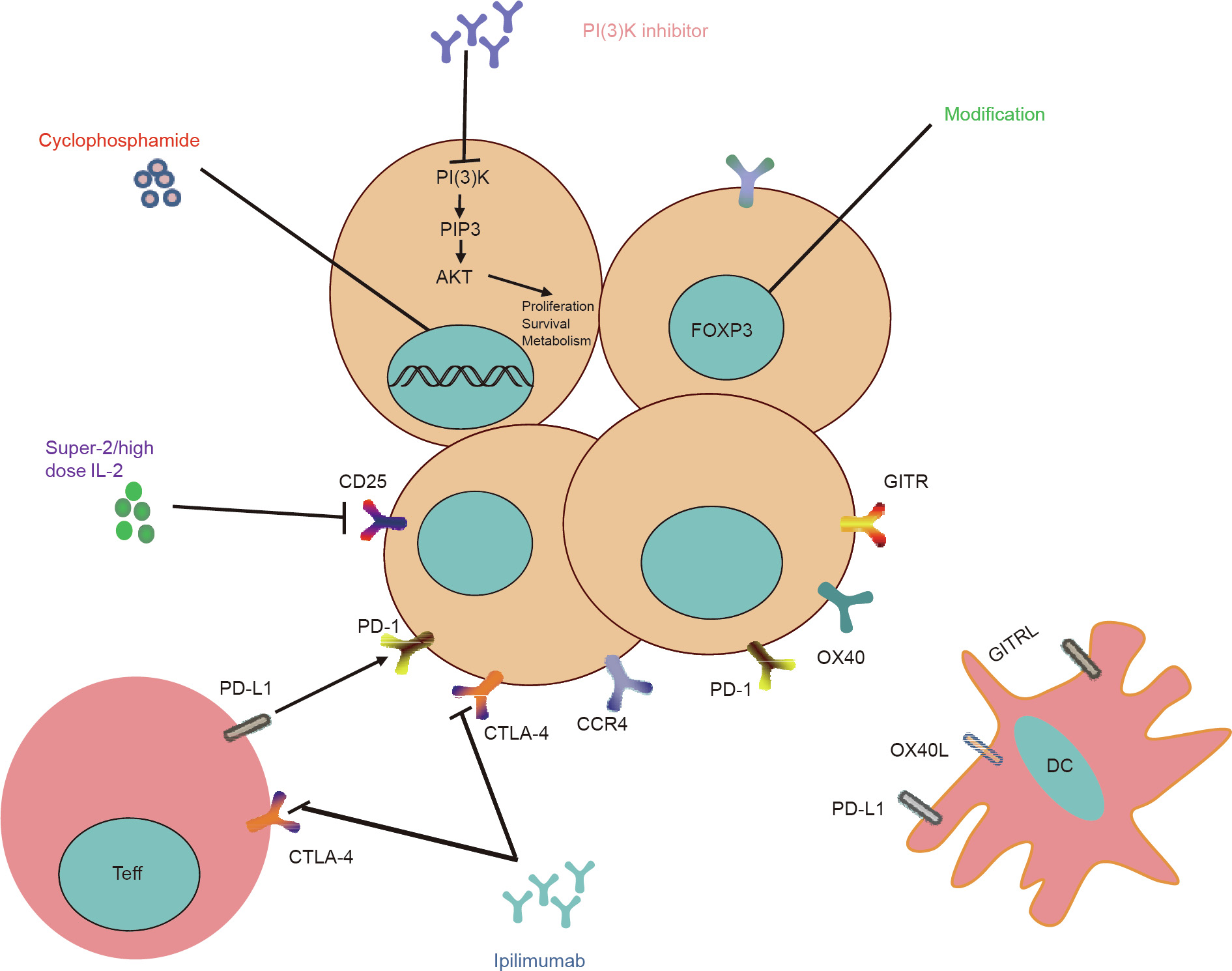
Fig. 2. The diverse function of Tregs in antitumor immunotherapy. PI(3)K: phosphatidylinositol 3-kinase; PIP3: phosphatidylinositol (3,4,5)-trisphosphate; OX40L: ligand for OX40; AKT: Akt serine/threonine kinase 1.
《7.2. Treg inhibitory function》
7.2. Treg inhibitory function
GITR and OX40 are co-stimulatory molecules that are constitutively expressed by Tregs. The agonistic antibodies of GITR and OX40 are capable of abrogating the suppressive function of Tregs, following the augmentation of Teff activity [91–94]. It is notable that when compared with the administration of anti-GITR mAb before or immediately after tumor inoculation, GITR treatment showed better results when tumor size had grown beyond a certain range [95]. Strategies targeting GITR and OX40 are now under clinical trials.
Except for surface molecules, signals that Tregs specifically depend on are potential targets that could be used to control Treg function. For example, phosphatidylinositol 3-kinase (PI(3)K) is a lipid kinase that reportedly plays a central role in cell proliferation, survival, and motility. Inactivation of PI(3)K has been shown to result in promising antitumor activity in human leukemia [96]. In a mice model, the blockade of PI(3)K has been shown to be capable of inducing the regression of various solid tumors [97].
An alternative strategy is to downregulate the expression of Foxp3, the key transcription factor of Tregs. Lentiviral Foxp3 shRNA has been used in mice for this purpose, and delivery led to restrained Treg function. Furthermore, the molecules involved in the posttranslational modification process of Foxp3 are also good targets. An additional concern is that inhibition of Foxp3 expression may evoke inescapable side effects. Therefore, the inhibition of Foxp3 expression requires further study.
《7.3. Specific Treg depletion》
7.3. Specific Treg depletion
CD25 is highly expressed by Tregs, while Teffs can transiently express CD25 after TCR stimulation. It has been generally acknowledged that the dual roles played by IL-2 in activating both Tregs and Teffs limit clinical use of the blockade of CD25 and IL-2. However, the potential to expand the utility of anti-CD25/IL-2 mAb has been demonstrated by various researchers. Levin et al. [98] have discovered that in comparison with IL-2, genetically modified IL2 ("super-2”) has a lower binding affinity to CD25+ cells and possesses the ability to induce a lower Treg expansion than CTLs; this finding indicates the promising clinical potential of ‘‘super-2”. A different IL-2 dose may also yield disparate results. In contrast with the disappointing results of low-dose IL-2 treatment, highdose IL-2 administration in metastatic melanoma and renal cell carcinoma patients increased people’s confidence in the application of IL-2 by yielding complete tumor regression in a fraction of patients and extending disease-free intervals in many other patients [99]. The role of anti-CD25 mAb is now controversial. The results of daclizumab (a humanized IgG1 monoclonal antibody targeting CD25) in combination with DC vaccination for the treatment of metastatic melanoma have been disappointing, due to the combined effects of the depletion of both Tregs and activated Teffs [100]. Interestingly, in breast cancer patients, the combination of daclizumab and vaccination produced favorable clinical responses without severe side effects [101]. Furthermore, anti-CD25 mAb administration in mice before they were inoculated with tumors was found to result in more effective antitumor responses than anti-CD25 mAb treatment after tumor inoculation [102].
Cyclophosphamide is an alkylating agent that is widely used in cancer chemotherapy. It can alkylate DNA, resulting in DNA crosslinking, and thus effectively kills proliferation cells. In mice bearing PROb cells (a cell line derived from a rat colon carcinoma), a single administration of cyclophosphamide induced the loss of a number of CD4+ CD25+ T cells, permitting delayed growth of PROb tumors [103]. High doses of cyclophosphamide severely affect all T cells, whereas low doses of cyclophosphamide over the long term selectively affect proliferating Tregs [104,105].
The removal of Tregs is often followed by severe autoimmune disorders because of the dual functions of Tregs: tumor immune escape and the maintenance of homeostasis. Thus, how specific Treg depletion can be achieved has become a key question that urgently requires a solution. CCR4 is a good target due to its specific expression on Tregs. Anti-CCR4 antibody is reported to have the capacity to selectively deplete Tregs and simultaneously increase the numbers of CD4+ and CD8+ T cells [106]. Moreover, overwhelming evidence has shown that chemokines and their receptors on Tregs are heterogeneous in various types of tumors. Examples include CCR8 in pancreatic cancer [107]; CCR4 with CCL22 in breast cancer; and CCR4 with CCL22, CCR10 with CCL28, and CXCR4 with CXCL12 in ovarian cancer [108]. The administration of these specific expressed chemokines may result in selective depletion of Tregs. It is noteworthy that the removal of Tregs cannot reactivate anergic T cells, which suggests that for the purpose of remodeling antitumor immunity, the depletion of Tregs and the re-priming of Teffs need to be combined [109].
《8. Conclusion》
8. Conclusion
FOXP3+ Treg is an important component of the mammalian immune system and plays a critical role in maintaining immune homeostasis. An increasing amount of data reveals that FOXP3+ Tregs may act as an effective suppressor of our immune system in order to kill tumor cells. In recent years, tumor immunotherapy that involves depleting Tregs-mediated immunosuppression has appeared to be effective in the clinic. In general, however, there is still a long way to go before it will be possible to cure the majority of human cancer patients using antitumor immunotherapy. For example, a very effective strategy has yet to be identified that can specifically deplete Tregs in tumors without causing harmful side effects. In the case of inflammation-driven tumor progression, the exact role of tumor-tissue-infiltrated Tregs and their functional plasticity and instability requires further study. More detailed functional studies are essential in order to deepen our understanding of tumor-tissue-infiltrated Tregs. Alternative approaches to analyze the living cells involved in the tumor microenvironment, such as the application of single-cell sequencing technology, are being explored; this newly emerging data will greatly promote our understanding of human tumors and assist in the development of new antitumor therapeutics.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Feng Xie, Rui Liang, Dan Li, and Bin Li declare that they have no conflict of interest or financial conflicts to disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




