
《1. Introduction》
1. Introduction
Thermodynamics forms the fundamental underpinning of reactivity, transformation, and stability, and controls processes such as synthesis, corrosion and degradation, environmental transport, catalysis, and biological reactivity. In the materials field, the wealth of new compounds, polymorphs, hybrid organic–inorganic hybrid materials and metal organic frameworks, high-entropy alloys, and multiphase and nanophase materials attained by a variety of non-equilibrium synthesis and processing methodologies has outrun the available thermodynamic data, hampering current understanding of synthetic pathways, materials compatibility, and longevity during use, degradation, corrosion, and dissolution, and limiting our understanding of environmental contamination and transport for new materials. In the geological and environmental sciences, similar needs exist for thermodynamic data for complex minerals. The excitement of new chemistry in planetary systems, both in our solar system and beyond, requires a broad-scale thermodynamic approach. The needs of materials science, earth and planetary science, and environmental science are both overlapping and complementary. Thus the boundaries between earth/planetary and materials science are increasingly porous.
At the same time, rapid developments in industry have resulted in an increasing need for improved materials, along with better ways to characterize them and study their properties, in order to explain different phenomena and process failure on a large scale. In this way, fundamental and applied thermodynamics are being brought ever closer together, making narrowly defined ‘‘pure science” a thing of the past and interdisciplinary studies, novel and hybrid materials, and broad collaborations across academia and industrial R&D the new future.
The capabilities of and interest in thermodynamic measurements have been on the rise again after years of decline. The commercial availability of a high-temperature Calvet calorimeter suitable for oxide melt solution calorimetry and related measurements [1,2] is a major advance, as are substantial improvements in the range, accuracy, and convenience of thermal analysis equipment and the ability to make accurate cryogenic heat capacity measurements on milligram-sized samples using commercial instruments [3]. These new capabilities, combined with the need for data for new materials, have resulted in the establishment of several new groups of younger researchers who are active in experimental thermodynamics, and especially in calorimetry.
These developments in experimental thermochemistry are paralleled by rapid progress in computational methods that integrate state-of-the-art calculations based on density functional theory (DFT), new simulation methods for characterizing energy, and free-energy landscapes. There are strategies for coupling DFT results and experimental data within the framework of free-energy modeling of phase diagrams and thermochemistry in complex multicomponent systems (e.g., the CALPHAD approach). New algorithms for automated high-throughput calculations, data mining, and machine learning lead to unprecedented new datasets and the integration of modern tools of data analytics. While these computational developments have progressed rapidly, in manycases their validation—and hence the establishment of the accuracy and predictive power of these methods—has been hindered by the lack of robust experimental thermodynamic data. At the same time, experimental techniques for elucidating details of the structures of solids at the molecular scale, nanoscale, and mesoscale have improved immensely, led by—but by no means limited to— experiments involving diffraction, scattering, and spectroscopy at synchrotron and neutron sources. Some examples of combined calorimetric, structural, and computational studies are provided in Refs. [4–7].
This triumvirate of experimental thermodynamics, structural investigations, and theory/computation is beginning to provide a completely new understanding of stability and reactivity in complex solids. Thermodynamics is undergoing a renaissance! The recently formed Thermodynamics Consortium↑ brings researchers together to further advance this combined broad and resurgent field.
↑ www.thermocon.org.
Refractory ceramic materials, whether of geologic, planetary, or technological origin, present unique challenges for thermodynamic measurements. Their unreactive nature, which is essential to their applications, is a stumbling block for equilibration and dissolution measurements. Their structures are complex and their compositions are often multicomponent. Non-stoichiometry, variable oxidation state, and order–disorder phenomena are common. In this paper I emphasize two topics: ① the development of new calorimetric instrumentation and techniques; and ② an example of the application of combined techniques to the complexity of order–disorder behavior in pyrochlore materials.
《2. Fundamentals of calorimetry》
2. Fundamentals of calorimetry
Calorimetry refers to the measurement of heat effects. It is often separated into two categories, although there is some overlap between them. The first category is measurement that does not intentionally change the chemical composition of the sample, but studies the absorption/release of heat as the sample temperature is changed, whether in a continuous scan, in small steps, or in large increments. These measurements provide the heat capacity, C, or its integral, the heat content (HT) or relative enthalpy (HT HTref), where T refers to temperature and Tref is a reference temperature, that is often taken as either room temperature or absolute zero (0 K). If the material undergoes a phase change on the timescale of the measurements, the enthalpy of the transition (fusion, crystallization, or solid state structural transition) is also measured. If the sample undergoes a spontaneous chemical change (e.g., dehydration, decomposition, or oxidation–reduction) during the measurements, this must also be considered in interpreting the measurements. The second category is reaction calorimetry, in which a chemical change is induced in the sample and/or its constituents to obtain a heat of reaction that can be used to calculate the heat of formation. This methodology can further be divided into two groups. The first group is direct reaction calorimetry, in which the products are formed rapidly from the reactants in the calorimeter (e.g., combustion calorimetry in oxygen [8] or fluorine [9] gas, or intermetallic compound formation from the elements at high temperature [10]). The second is solution calorimetry, in which reactants and products synthesized outside the calorimeter are then each dissolved in a suitable solvent to produce the same final state; the difference in the enthalpy of solution of the reactants and products gives the enthalpy of reaction [2,11,12]. The solvent can be an aqueous system near room temperature (e.g., aqueous hydrochloric or hydrofluoric acid), or a molten metal or molten salt at high temperature. For refractory ceramics and minerals, solution calorimetry using molten oxide solvents at 700– 800 °C has developed over the last several decades into a wellestablished and very powerful technique [2].
《3. Cryogenic heat capacity measurements and standard entropies》
3. Cryogenic heat capacity measurements and standard entropies
To obtain the standard entropy of a crystalline material at room temperature, ST, it is necessary to integrate the heat capacity from a temperature as close as possible to absolute zero to the temperature of interest. Thus cryogenic heat capacity measurements down to the temperature of liquid helium (4 K), or even lower, are essential. Such measurements were traditionally done by adiabatic calorimetry, using large custom-built equipment and samples of 10–100 g. With the advent of more modern instrumentation in the 1980s and 1990s, sample sizes were reduced [13–16], but the labor requirements to perform the measurements remained very demanding.
In the last three decades, thermal relaxation techniques were developed in a variety of laboratories that significantly reduced sample sizes to the milligram and even microgram level [17]. Hellman et al. [18,19] further refined the thermal relaxation technique to measure thin films and powders. This method was successfully applied to a powdered sample of the high-pressure spinel form of fayalite, Fe2SiO4 [20].
Quantum Design, Inc. developed and has sold for some time the Physical Property Measurement System (PPMS) featuring a heat capacity option utilizing the thermal relaxation method. The PPMS heat capacity option is highly automated and can generate acceptable third-law quality heat capacity data in the temperature range of 2–300 K using sample masses of less than 10 mg. However, the standard PPMS method initially worked well (i.e., with high accuracy) only for highly conductive metallic samples. Over the past decade, Shi et al. [3] have developed sample mounting techniques that have eliminated errors for poor thermally conducting samples. Parallel developments have been achieved by Dachs and Benisek [21]. The next frontier for the PPMS is to develop sample platforms accessible to the general scientist that decrease sample sizes to the 100 μg level while maintaining acceptable accuracy and resolution. This capability will be necessary in order to measure the heat capacity of quenched high-pressure phases and other metastable materials.
《4. Thermal analysis》
4. Thermal analysis
Methods of differential thermal analysis (DTA) and differential scanning calorimetry (DSC) [22] are rooted in the measurement of sample temperature heating or cooling. Heat flow traces correspond to differences in temperature between a sample and a reference crucible. This methodology is well developed and widely used for biological materials, polymers, and ceramics up to 1600 °C. DTA instruments for operation up to 2400 °C are available from several manufacturers. Quantitative measurements of heat capacities and enthalpies of phase transitions (both in the solid state and melting) are achievable when proper care is taken and appropriate calibration standards are available.
Recently, we further developed the DTA method to enhance its potential to provide data on fusion and phase transition enthalpies above 1600 °C. A DTA (Setaram Setsys 2400, Caluire, France) equipped with a graphite furnace and W-WRe sensor was modified to allow operation up to 2500 °C and the measurement of sample temperatures by radiation thermometry (Fig. 1) [23]. Enthalpies of phase transitions of HfO2 [7] and La2O3 [24] and fusion enthalpies of La2Zr2O7 [25], LaAlO3 [26], La2O3 [27], and Nd2O3 [27] were successfully measured with samples sealed in tungsten crucibles to prevent reactions with the carbon present in the furnace. The recent independent measurement of the fusion enthalpy of Y2O3 [6] makes high-temperature sensitivity calibration possible, which extends DTA capabilities to provide thermodynamic data above 2000 °C.
《Fig. 1》
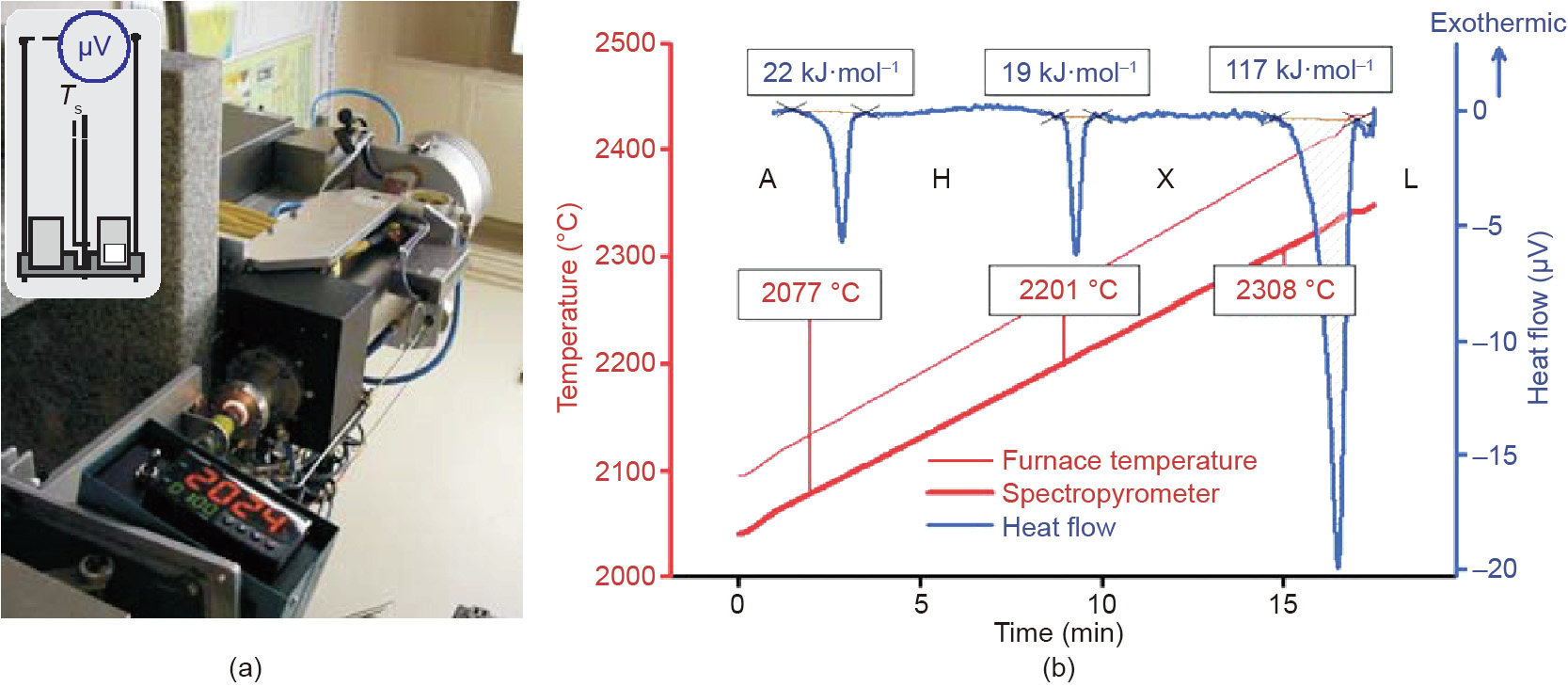
Fig. 1. (a) High-temperature DTA apparatus and (b) phase transitions in Nd2O3. Ts is the temperature of the sample. A (trigonal), H (hexagonal), and X (cubic) indicate the structures of the solid state phases, while L indicates the liquid state. Reprinted from Ref. [23] with permission of The American Ceramic Society, ![]() 2017.
2017.
《5. Ultra-high-temperature drop calorimetry》
5. Ultra-high-temperature drop calorimetry
DSC above 2000 °C has two limitations. The first is the intrinsic reactivity of the crucible materials and the sample at very high temperatures, which is almost impossible to avoid. The second limitation is the lack of reliable DSC standards (i.e., materials with known melting points and heats of fusion) at temperatures higher than the melting point of alumina (Al2O3). Considering how to ameliorate these problems led Ushakov and Navrotsky to develop a new type of calorimeter for work up to 3000 C or perhaps higher. Their ‘‘drop-and-catch” (DnC) calorimeter [23,26–28] is a miniaturized drop calorimeter in which a hot sample is dropped into a room temperature calorimeter and its heat content is measured. Earlier drop calorimeters used large samples (tens to hundreds of grams) contained in noble metal or tungsten containers in a furnace. Thus, sample-crucible reactivity was still a potential problem (albeit minimized in principle by a large sample having a relatively small surface-volume ratio). However, the large sample size meant that the sample in the calorimeter cooled relatively slowly, leading to possible heterogeneity in the final product. The DnC overcomes these difficulties by using a small sphere of aerodynamically levitated sample heated by a laser and then dropped a short distance into a sensitive room temperature calorimeter with copper plates to catch the sample and cool it rapidly and reproducibly. This process is shown schematically in Fig. 2 [23], which also shows calorimetric data for alumina, clearly indicating its heat of fusion. The DnC has been applied to several rare earth oxides and to zirconia and hafnia [6,7,24]. The current state of the art is that heats of fusion can be measured with an accuracy of 5%–10%. Accurate heat capacity determination from the slope of the curve of heat content versus temperature is being developed; the main issues to be overcome are accurate temperature measurement and the control and modeling of temperature gradient within the levitated sample.
《Fig. 2》

Fig. 2. DnC calorimeter (a) schematic, (b) operation, and (c) enthalpy versus temperature curve for Al2O3. Diameter of sample beads is 2.5–3 mm. HF: heat flow. Reprinted from Ref. [23] with permission of The American Ceramic Society, ![]() 2017.
2017.
《6. Solution calorimetry》
6. Solution calorimetry
In solution calorimetry, reactants and products are each dissolved in a solvent to form a final solution of identical concentration; this makes it possible to determine the heat of reaction as the difference in their heats of solution. However, since most crystalline oxides, nitrides, and oxynitrides are refractory (a desirable characteristic), they do not dissolve readily in aqueous solutions. Instead, a high-temperature molten salt solvent is used in a specialized Calvet-type heat flow calorimeter [2] (Fig. 3).
《Fig. 3》

Fig. 3. Schematic of high-temperature Calvet-type heat flow calorimeter for oxide melt solution calorimetry and sample assembly.
Dissolution reactions are commonly carried out by dropping small pellets (1–5 mg) at room temperature into 10–20 g of solvent—typically lead borate (2PbO–B2O3) or sodium molybdate (3Na2O–4MoO3)—at 700 or 800 °C (a so-called drop solution experiment). Measured enthalpies of drop solution have been summarized for a large variety of oxides [2]. These values are reliable (with uncertainties of ±0.5% to 2%, depending on the chemistry) and have been shown to remain constant when done on different calorimeters over a timespan of years. The lead borate solvent is optimized for studying silicates, while the sodium molybdate solvent is generally better for oxides such as ZrO2, TiO2, and the rare earth and actinide oxides.
Recent developments in calorimetric techniques include bubbling gas through the calorimetric solvent to control oxygen fugacity and to stir the melt in order to enhance dissolution rates and prevent local saturation. Because the Mo6+ in sodium molybdate is easily reduced to Mo5+, this solvent acts as an excellent catalyst for the oxidation of nitrides, carbides, chalcogenides, and metals upon dissolution, providing reliable thermodynamic cycles for determining their heats of formation [2]. A very recent development is oxidative drop solution calorimetry of metals, which enables the determination of heats of formation of a variety of alloys (in preparation).
When oxide melt solution calorimetry was first developed, it required samples of about 100 mg per run. We now routinely work with samples of 1–3 mg. Further miniaturization may be possible, although it is unlikely that samples smaller than about 0.1 mg could be studied.
The commercialization of Navrotsky’s calorimeter as the Setaram AlexSYS [1,2] has made high-temperature oxide melt solution calorimetry and related techniques accessible to a wide range of users. About 15 such calorimeters have been bought and established in new laboratories worldwide, with new groups working on problems related to metals, batteries, nuclear waste, and other topics. This development has transformed a seemingly esoteric and specialized technique into one that is accessible to independent users. Publications are starting to emerge from these new groups; this activity is part of the renaissance of thermodynamics mentioned in the introduction.
《7. Disorder on several length scales—Pyrochlores as an example》
7. Disorder on several length scales—Pyrochlores as an example
Complex oxides can be defined as oxides containing a variety of cations (and sometimes anions) distributed over several different types of sites (sublattices). The term ceramics generally refers to refractory materials with high melting points. Complex ceramics are used extensively in applications ranging from thermal barrier coatings to multiferroics to nuclear waste immobilization. Igneous minerals fall into the class of high-temperature ceramic materials.
The equilibrium distribution of ions, vacancies, and oxidation states depends on composition, temperature, and pressure. In addition, a wealth of out-of-equilibrium states can be accessed using various techniques: quenching from high temperature or pressure, low temperature aqueous or solvothermal synthesis of disordered nanomaterials, vapor deposition, grinding, and radiation damage. These metastable states can sometimes persist indefinitely under ambient conditions and—perhaps surprisingly—at higher temperatures as well. The physical properties (e.g., ionic and electronic conductivity), thermodynamic properties (e.g., phase stability and aqueous solubility), and reactivity (e.g., rates of phase transformation, corrosion, and dissolution) all vary with structural state. Such variations can be deleterious to function, but can also be harnessed to provide desirable properties such as higher ionic conductivity. The example of pyrochlores, discussed below, points to the richness and variability of structural state and metastability as a function of preparation conditions, and suggests studies to determine the energetics of the underlying order–disorder phenomena. It also shows the interplay of calorimetric, structural, and computational approaches. Because different structural states are often (though not always) close in energy, they define an ‘‘energy landscape” over which reactions can proceed, sometimes finding local minima, and eventually global minima, in free energy along various reaction pathways.
Pyrochlores have an idealized general formula of A2B2O7, where A is tetravalent and B is trivalent. They exhibit a cubic unit cell, which can be considered a 2 × 2 × 2 superstructure of the fluorite unit cell, with the A- and B-site cations being eight- and six-fold coordinated with oxygen, respectively. Deviations from the simple fluorite structure (general formula AO2) include the presence of two distinct cation species arranged in an ordered manner over the cation sublattice and the replacement of one eighth of the oxygen anions by vacancies for charge balance [29].
Many pyrochlores undergo an order–disorder transformation at high temperatures or under irradiation, in which the pyrochlore superstructure is lost, leaving only fluorite-type long-range order [29–34]. Neutron total scattering experiments, however, reveal that the cations and oxygen atoms in the materials are not randomly arranged at the atomic level, but only appear so when sampling over longer scales [29]. The short-range order occurring in nanoscale domains has been described as similar to that in the weberite structure. Radiation damage can also produce X-ray amorphous samples that nevertheless contain weberite-like nanodomains [29].
Much of our recent work on pyrochlores has been supported by the US Department of Energy’s Energy Frontier Research Center— Materials Science of Actinides, because of the importance of pyrochlores as possible nuclear waste ceramics. However, the ubiquitous family of pyrochlore materials has many other applications, ranging from oxide dispersion strengthened steels to electronic, multiferroic, and dielectric materials. As part of a study on pyrochlores damaged by swift heavy ion irradiation and by grinding, our collaboration performed total neutron scattering studies of short-, mid-, and long-range order, computational work, and calorimetry [30–33]. We used calorimetry to determine the energetic differences among radiation-damaged, annealed, and undamaged Dy2Ti2O7, and the heat release during damage annealing [33]. The damage recovery was shown to be a multistep process that is much more complex than previously thought. It involves decoupled long-range recrystallization and short-range defect recovery operating at different temperatures and timescales (Fig. 4) [33]. In particular, a short-range order analogous to that in weberite, where there is more anion disordering than in pyrochlore but less than in a randomized defect fluorite structure, has been found by neutron diffraction in radiation-damaged pyrochlores that appear to be either defect fluorite or amorphous to laboratory X-ray diffraction (XRD) [29].
The long-range amorphous-to-crystalline transformation occurs relatively quickly within a narrow temperature regime (800– 900 °C), but accounts for only about one third of the overall heat release found by oxide melt solution calorimetry as the difference in the enthalpy of solution of fully ordered pyrochlore and amorphized samples. The local reordering from weberite-like order into pyrochlore-like order is much more gradual and operates mainly between 900 and 1200 °C, releasing an additional third of the total stored energy. Most interestingly, the sample does not fully recover at 1200 °C, either structurally or energetically, even after heating well above the initial recrystallization temperature. A large fraction of the total energy of amorphization is still stored in the sample, even though it appears to be fully recovered in terms of the long-range structure detected by XRD. This remaining stored energy can be mainly linked to the short-range weberite-like ordering that persists, as seen in neutron studies, even after hightemperature annealing. These relationships are shown schematically in Fig. 4. The retention of the local defects and stored energy due to incomplete damage recovery at elevated temperatures will affect physical and transport properties, leaching behavior, and radionuclide immobilization.
《Fig. 4》

Fig. 4. Schematic diagram showing enthalpy differences among ordered, disordered, and amorphized dysprosium titanate. The red symbol shows the inability of the sample to return to the fully ordered state on annealing for several hours to 1200 °C.
Pyrochlores also undergo disordering at high temperature. Indeed, their occurrence, controlled by their enthalpies of formation [35–37], is limited to the lighter (larger) rare earths, with the defect fluorite structure being stable for the heavier rare earths. Using high-temperature thermal analysis, we determined the enthalpy of the reversible pyrochlore-to-defect-fluorite transition for several pyrochlores [34]. The calculated entropy changes at the transition are consistent with values calculated from the configurational entropy change on disordering.
Other pyrochlores disorder gradually with increasing temperature. Using in situ synchrotron diffraction, we traced the temperature dependence of both cation and anion disordering [34]. We applied a simple thermodynamic model, analogous to that used for spinels [38], to calculate interchange enthalpies from the observed equilibrium distributions. The cation interchange enthalpies to form antisite defects depend on the nature of the rare earth, while the oxygen vacancy disordering enthalpies to form Frenkel pairs show little composition dependence. The enthalpy values obtained follow trends predicted by ab initio calculations [39].
《8. Conclusions and prospective research》
8. Conclusions and prospective research
Many outstanding problems in materials, geochemistry, and planetary science will benefit from the advances in and unification of thermodynamic measurements, structural studies, and computational approaches. Applications include the structure of planetary interiors and materials under extreme conditions, environmental science and pollution control and abatement, new materials for catalysis and energy applications, and ultra-high-temperature ceramics and coatings, to name a few. In each case, the thermodynamics involve both understanding synthesis, persistence, and transformation, and constraining fundamental properties at the molecular, nanoscopic, mesoscopic, and macroscopic length scales. As the capabilities grow and new researchers—especially young scientists—are drawn into thermodynamics, this field will expand, thrive, and continue to prove its usefulness.
《Acknowledgements》
Acknowledgements
The work reviewed here received support over many years from the US Department of Energy and the National Science Foundation, United States. Specifically, the work on pyrochlores was funded by the Materials Science of Actinides, an Energy Frontier Research Center funded by the US Department of Energy, Office of Science, Basic Energy Sciences (DE-SC0001089), while that on ultra-hightemperature calorimetry was supported by the National Science Foundation DMR (1506229 and 1835848).











 京公网安备 11010502051620号
京公网安备 11010502051620号




