

《1. Introduction》
1. Introduction
Salvia Linn. (S. Linn.) is a large genus of Labiatae that comprises approximately 1000 species worldwide; of these species, 84 are found in China—mainly in Southwest China [1]. Among these 84 species, nearly 30 have been recorded as medicinal plants [2]. Danshen is a representative traditional Chinese medicine (TCM) [3] that has a significant effect on the treatment of cardiovascular and cerebrovascular diseases [4,5]. The Chinese Pharmacopoeia indicates that S. miltiorrhiza Bunge is the only source of Danshen [6]. However, many other Salvia plants have similar chemical components and corresponding pharmacological effects to those of Danshen, and are used as Danshen in nongovernmental applications and local medication. For example, S. przewalskii is an herbaceous perennial plant that is widely distributed in Southwest China [7] and has been used as a substitute for S. miltiorrhiza for at least 300 years. This plant is also used as a substitute for Gentiana macrophylla in Sichuan Province. Many studies have shown that S. przewalskii has similar components to those of S. miltiorrhiza ; however, the amount of fat-soluble components in the root of S. przewalskii is several times higher than in S. miltiorrhiza, giving S. przewalskii great application value [8–10]. S. bulleyana, also known as “purple Danshen” is mainly distributed in Dali in Yunnan Province and is often used as Danshen folk medicine [11]. However, the chloroplast (cp) genomes of most Salvia plants remain unknown.
Chloroplasts, which play a key role in autotrophic photosynthesis, are important organelles in green plants [12–14]. The cp genome is typically a circular multicopy DNA molecule in cells [15,16]. The structure of plant cp genome is conservative and can be divided into four segments—that is, two copies of inverted repeat (IR) regions (IRa and IRb), which are separated by a large single-copy (LSC) region and a small single-copy (SSC) region [17,18]. The sizes of the cp genomes of different species differ mainly due to IR contraction or expansion [19,20]. With the advent of new sequencing technologies, sequencing is becoming faster and cheaper; more than 2000 cp genomes have now been reported to the National Coalition Building Institute (NCBI) [21]. At present, the use of a DNA barcode to differentiate between closely related species and infraspecific taxa is hampered due to short gene fragments and a low number of phylogenetic informative sites. Therefore, some researchers have proposed using the whole cp genome as a ‘‘super barcode” for species identification [22]. Given its high expression efficiency, site-specific integration, and maternal inheritance, cp transformation techniques have shown considerable potential for genetic improvement [23–26].
Genomic data, including organelle genomic data, can provide a molecular basis for the study of the original species of TCM. In our previous work, we have studied many genome sequences [27–29], including the complete cp genome sequence of S. miltiorrhiza in 2013 [30] and the draft sequence and analysis of the S. miltiorrhiza genome in 2016 [31]. In the present study, we obtained the complete cp genome sequence of S. przewalskii and S. bulleyana using a next-generation sequencing platform. We then compared the two cp genomes with those of the two other Salvia spp. that have been reported, in terms of genome organization, repeat sequence, and IR length. Finally, we performed a phylogenetic analysis based on the whole cp genome of 16 angiosperms. These efforts provide additional information for constructing the cp genome library of Salvia, which will aid in identifying Salvia spp. and provide insights into its evolutionary origins.
《2. Results》
2. Results
《2.1. Cp genome organizations》
2.1. Cp genome organizations
Raw data (approximately 5.9 × 109 and 1.372 × 1010, respectively) and trimmed data (approximately 5.2 × 109 and 1.343 × 1010, respectively) were obtained from S. przewalskii and S. bulleyana. The resulting cp genomes of the two Salvia spp., S. przewalskii (MH603953) and S. bulleyana (MH603954), have been presented to the NCBI. The total cp genome sizes of the two Salvia spp. are 151 319 bp and 151 547 bp, respectively. Regarding the cp genomes of the two Salvia spp., both S. przewalskii and S. bulleyana have a typical quadripartite structure, like the majority of flowering plants, composed of a pair of IRs (50 982 and 51 098 bp) and two single-copy regions (LSC: 82 732 and 82 853 bp; SSC: 17 605 and 17 596 bp), as shown in Fig. 1 and Table 1.
《Fig. 1》

Fig. 1. Gene map of the S. przewalskii and S. bulleyana cp genomes. The genes inside and outside the outer circle are transcribed in the direction of the grey arrows inside and outside at the top. Genes are classified into 14 groups according to their biological function and are shown by different colored boxes. Within the inner circle, dark grey represents GC content and light grey represents AT content.
《Table 1》
Table 1 Summary of the base composition of the cp genomes of four Salvia species.

The cp genome of S. przewalskii is the shortest of the four sequenced Salvia spp., while S. bulleyana ranks between S. miltiorrhiza and S. japonica in length. The cp genome lengths of the four species range from 151 319 to 153 995 bp, while the LSC region lengths range from 82 695 to 84 573 bp. The IR region ranges from 50 982 to 51 832 bp, and the SSC region ranges from 17 555 to 17 605 bp. The GC content of the S. przewalskii and S. bulleyana cp genomes is 37.96% and 37.99%, respectively, while the IR regions possess a higher GC content (43.11% and 43.12%) than the LSC (36.08% and 36.12%) and SSC regions (31.88% and 31.89%). In general, the IR region has four ribosomal RNA (rRNA) genes enriched in GC [32]. The S. przewalskii and S. bulleyana cp genomes have more AT than GC; the same is true for the cp genomes of the two other Salvia spp., and for those of other land plants [33–36].
In this study, we annotated 134 genes in two Salvia cp genomes, of which 114 are unique, consisting of 80 protein-coding, 30 transfer RNA (tRNA), and four rRNA genes (Fig. 1, Table S1 in Supplementary data (SD)). Out of these, 18 genes, consisting of seven protein-coding, seven tRNA, and four rRNA genes, are repetitive in the IR regions. A total of 61 protein-coding and 22 tRNA genes are present in the LSC region, whereas 12 protein-coding and one tRNA genes are present in the SSC region. One ycf1 pseudogene and one rps19 pseudogene are located in two IR boundary regions (Fig. S1 in SD).
The quantity and type of genes in the two Salvia cp genomes are the same as those in the two other Salvia spp. A slight variation was observed in the nucleotide composition in the coding sequence (CDS) of the four Salvia cp genomes (Table 2). The GC contents are 45.75%/44.91%, 38.19%/38.49%, and 30.32%/30.89% at the first, second, and third codon positions in two Salvia CDS regions identified in this study, respectively (Table 2). The contents of the other two Salvia spp. are 45.77%/45.73%, 38.21%/38.13%, and 30.38%/30.17% at the first, second, and third codon positions, respectively. The trait of a preference for AT the third codon position that was observed in the cp genomes of these four Salvia species also appears in other angiosperms [37–40].
《Table 2》
Table 2 Gene number and CDS nucleotide composition of the cp genomes of the four Salvia species.

A total of 87 protein-coding genes have 26 439 and 26 432 codons in the S. przewalskii and S. bulleyana cp genomes, respectively, compared with 26483 and 26485 in S. miltiorrhiza and S. japonica, respectively (Table S2 in SD). Out of these codons, AUU (1096/1106) encoding isoleucine (Ile) and UGC (76/114) encoding cysteine are the most and least used in S. przewalskii and S. bulleyana, respectively. AUU and UGC are also the most and least used in the two other Salvia cp genomes (1100/1102 and 70/71). The relative synonymous codon usages (RSCUs) of the four cp genomes differ slightly from each other. Multiple codons are present for all the amino acids, in addition to methionine and tryptophan (Fig. 2). There are six synonymous codons for arginine (Arg), leucine (Leu), and serine (Ser); four synonymous codons for each of valine (Val), proline (Pro), threonine (Thr), alanine (Ala), and glycine (Gly); three synonymous codons for each of Ile and stop codons; and two synonymous codons for each of the remaining amino acids. The synonymous codon is usually mutated only at the third position, thereby reducing harmful mutations.
《Fig. 2》
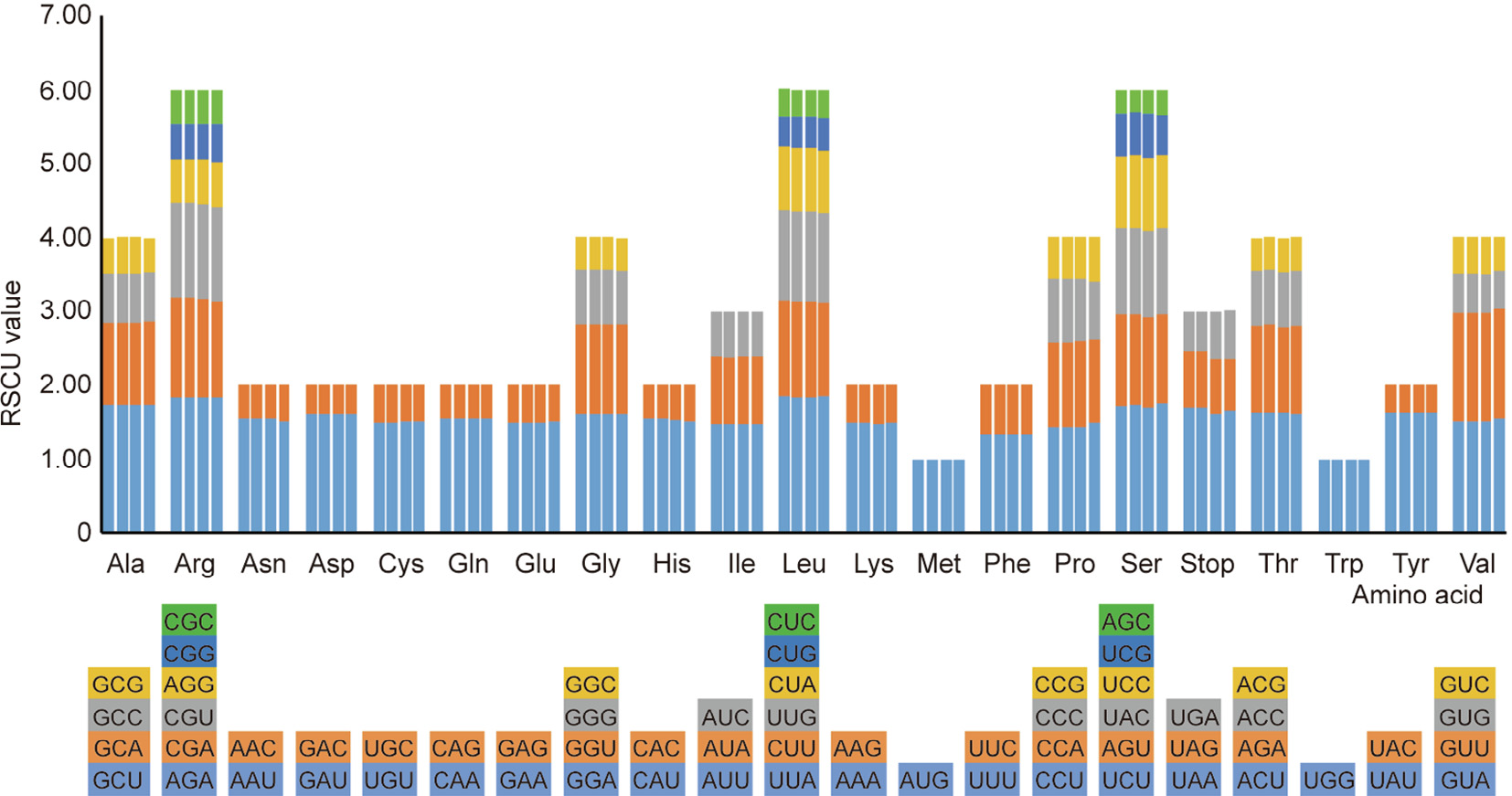
Fig. 2. Codon content for the CDS in the four Salvia cp genomes. The abscissa represents 20 amino acids and terminators, while the ordinate represents the RSCU value. For each amino acid, the corresponding species from left to right are S. przewalskii, S. bulleyana, S. miltiorrhiza, and S. japonica. The different colors of each amino acid correspond to the codon of the same color below. Asn: asparagine; Asp: asparticacid; Cys: cysteine; Gln: Glutarnine; Glu: glutamicacid; His: histidine; Lys: lysine; Met: methionine; Phe: phenylalanine; Trp: tryptophan; Tyr: tyrosine.
Introns play a crucial part in the regulation of gene expression and can enhance exogenous gene expression at plant sites to produce ideal agronomic traits [41,42]. A total of 18 genes containing introns were observed in the four Salvia cp genomes—15 containing one intron and three containing two introns. The intron of the trnK-UUU gene is the longest, and contains the matK gene. The 5’ end of the rps12 gene is located in the LSC region, while the 3’ end is located in the IR regions, making it a trans-spliced gene. In general, the exon lengths are conserved in the four Salvia spp., except for the ndhB gene (Table S3 in SD).
The 18 genes of the four Salvia spp. were compared with those of four other Labiate species—namely, Mentha longifolia, Ocimum basilicum, Perilla frutescens, and Scutellaria baicalensis. Minor variations were observed in most exon lengths of the eight Labiate cp genomes; however, some of these genes are only conserved in Salvia spp. and Mentha longifolia. These genes include trnV-UAC, rpoC1, and ycf3, where the intron phase are different in the eight Labiate cp genomes. The exon length of the rps16 gene is specific for the four Salvia spp. Greater variation in the intron length was observed than in the exon length, although the intron lengths of some genes, such as trnL-UAA and rpl2, are the same in three of the four Salvia spp., with the exception of S. japonica.
RNA editing participates in plastid transcription regulation, which can enrich transcription and protein diversity [43–45]. In this study, 35 genes of the four Salvia cp genomes were predicted for their potential RNA editing sites. A total of 43 RNA editing sites were predicted; of these, 37 are common sites of the four species (Table S4 in SD). Of the 35 genes, 16—including atpA, atpB, clpP, petD, petG, petL, psaB, psaI, psbB, psbE, psbF, psbL, rpl23, rpoC1, rps8, and ycf3—were not measured for their potential RNA editing sites. The rps16 gene was not measured for its potential RNA editing sites in S. japonica, but one potential RNA editing site was observed in the three other species. Of the 43 potential RNA editing sites, 11 were observed at the first position of the corresponding codon and 32 were observed at the second position. No potential RNA editing site was observed at the third position, and the base conversion type is all C to T. This result is similar to those of other land plants [46,47]. The conversion of amino acids from Ser to Leu occurs most frequently, while the conversions from Pro to Ser and Thr to Ile occur least frequently.
《2.2. Repeat and simple sequence repeat (SSR) analyses》
2.2. Repeat and simple sequence repeat (SSR) analyses
In the repeat sequences analysis, 43 repeats—comprising 21 forward and 22 palindromic repeats—were found in both Salvia spp. by using REPuter (Table S5 in SD, Fig. 3). However, most repeats were found in the ycf2 coding region; four were found between the ycf3 intron region and the intergenic spacer (IGS) of rps12 and trnV-GAC, and four were found in the IGS of rrn4.5 and rrn5 . In S . przewalskii and S . bulleyana, 17 and 21 tandem repeats were detected, respectively (Table S6 in SD, Fig. 3). Approximately half of these repeats are located in the ycf1 and ycf2 genes, while the other half of the repeats are located in the IGS regions. The two longest repeats, which are approximately 90 bp in length, are present in ycf2 in the two Salvia spp.
《Fig. 3》

Fig. 3. Repeat sequences analysis of eight cp genomes. (a) Repeat types in eight cp genomes; (b) tandem repeats in eight cp genomes; (c) forward repeats in eight cp genomes; (d) palindromic repeats in eight cp genomes. In (a), different colors show different repeat types; in (b–d), different colors show different lengths. The ordinate represents the number of repeats; in the abscissa, numbers 1–8 represent the following: 1 for S. przewalskii, 2 for S. bulleyana, 3 for S. miltiorrhiza, 4 for S. japonica, 5 for Mentha longifolia, 6 for Ocimum basilicum, 7 for Perilla frutescens, and 8 for Scutellaria baicalensis.
A comparative analysis of the repeats in the eight Labiate cp genomes showed that S . przewalskii and S . bulleyana are resemblant with the other cp genomes in repeat type, while S . japonica possesses more long-segment repeats than the others. The lengths of the tandem repeats in the eight Labiate cp genomes range from 10 to 30 bp, while the lengths of the forward and palindromic repeats mostly range from 30 to 45 bp.
SSRs are widely distributed at different locations in the genome [48]. The cp genome has the characteristics of uniparental inheritance, and SSRs have a high variation level within the same species. Thus, cp SSRs have been widely used as molecular markers in the study of genetic map construction, target gene calibration, and mapping [49–51]. Here, a total of 178 SSRs, comprising 134 mononucleotide (mono), 35 dinucleotide (di), seven tetranucleotide (tetra), and two pentanucleotide (penta) SSRs were observed in the S . przewalskii cp genome; a total of 177 SSRs, comprising 136 mono, 32 di, and nine tetra SSRs were observed in the S. bulleyana cp genome (Fig. 4 , Table S7 in SD). The major type of SSR is the mono SSR; the A/T type SSR (132 129) accounts for the vast majority of the mono SSRs.
《Fig. 4》

Fig. 4. SSR analysis of eight cp genomes. The ordinate represents the number of SSRs.
A comparison of the SSRs in the eight cp genomes showed that all eight cp genomes are similar. Most of the SSRs in the eight cp genomes consist of mono and di repeat motifs. The mono repeats vary from 109 (Ocimum basilicum) to 136 (S. bulleyana), and the di repeats vary from 25 ( S . miltiorrhiza) to 39 (Perilla frutescens). However, three or more oligonucleotide repeats are relatively low in number but rich in types (Table S7).
《2.3. Synteny comparison and SNP analyses》
2.3. Synteny comparison and SNP analyses
The pairwise cp genomic alignment of S . przewalskii and S . bulleyana with the two other Salvia cp genomes was conducted with annotated S . przewalskii cp genomes as a reference, using mVISTA (Fig. S2 in SD). The variable sites of the cp genomes and the 80 unigenes of the four Salvia cp genomes were analyzed (Fig. 5 and Table S8 in SD). The comparison and single-nucleotide polymorphism (SNP) analyses of the alignment of the four cp genomes revealed that the IR regions are more conserved than the LSC and SSC regions, in which divergences are dispersed. The noncoding regions have more divergent regions than the coding regions. In these cp genomes, highly divergent regions appear in the IGSs, including rps16–trnQ, trnG–trnS , atpH–atpI , psbA–ycf3 , ycf4–cenA and thrnV–ndhC in the LSC region and ycf1–rps15 , rpl32–trnL, and ndhI–ndhG in the SSC region. Some divergences were also observed in the coding regions of the rpoC2 , ndhF, and ycf1 genes in the four cp genomes. S . przewalskii and S . bulleyana are more similar to S .miltiorrhiza than to S . japonica .
《Fig. 5》
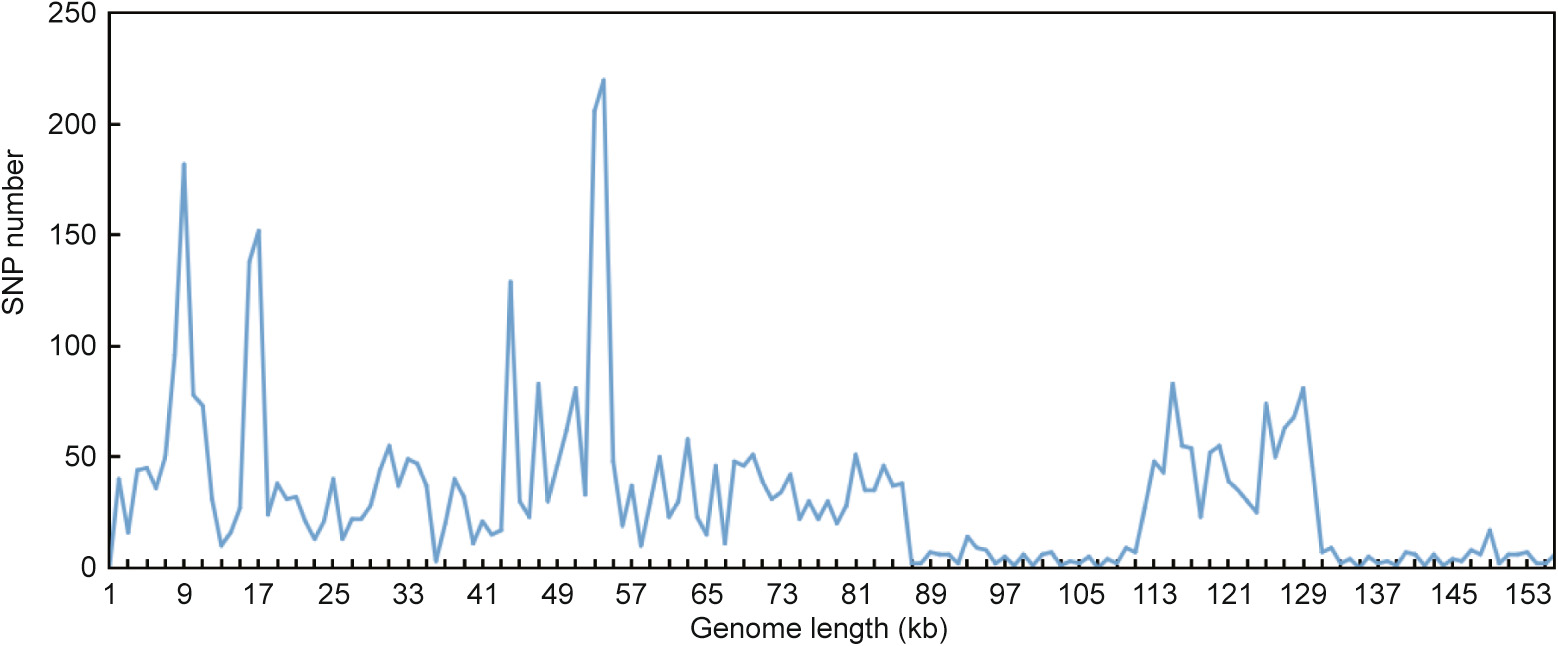
Fig. 5. Statistics of the SNPs in the four cp genomes.
《2.4. Phylogenetic analysis》
2.4. Phylogenetic analysis
A phylogenetic tree was built based on eight Labiate, four Solanaceae, and three other Asterales cp genomes using the maximum likelihood (ML) method, with Arabidopsis thaliana as the outgroup (Fig. 6). All of these have a bootstrap value of 100%, and in the ML tree, all four Salvia spp. form a robust monophyletic branch. Two Salvia spp.—namely, S. przewalskii and S. bulleyana—are clustered together in one terminal branch, thereby representing subgen. Salvia. All eight Labiate spp. are clustered together in one monophyletic group and nested in the branch of Sesamum indicum (Pedaliaceae), Boea hygrometrica (Gesneriaceae), and Olea europaea (Burseraceae). This result is consistent with the findings of a previous report [30].
《Fig. 6》

Fig. 6. ML phylogenetic tree reconstruction containing the cp genomes of 16 plants. Arabidopsis thaliana was set as the outgroup.
《3. Methods》
3. Methods
《3.1. Plant material and DNA extraction》
3.1. Plant material and DNA extraction
S. przewalskii was collected from Yulong County in the Lijiang City in Yunnan Province, and S. bulleyana was collected from Wulongba, Binchuan County in the Dali Bai Autonomous Prefecture in Yunnan Province. The total genomic DNA of S. przewalskii was extracted from 100 mg of fresh leaf using the DNeasy Plant Mini Kit (QIAGEN GmbH, Germany), while the total genomic DNA of S. bulleyana was extracted from 100 mg of fresh leaf using the modified cetyl trimethyl ammonium bromide (CTAB) method [52]. The quality and concentration of the genomic DNA were estimated using agarose gel electrophoresis and a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific Inc., USA). Qualified DNA was used for library construction in terms of the user guide. The Illumina HiSeq 1500 platform (Illumina Inc., USA) was used for sequencing.
《3.2. Assembly and annotation of the two Salvia spp.》
3.2. Assembly and annotation of the two Salvia spp.
The whole-genome sequences were used to extract the cp genomes. First, raw reads were evaluated using FastQC and trimmed using Trimmomatic to remove low-quality bases (Q < 30, Q = -10 log10(error P), Q < 30 means that limiting the error rate < 0.001, length < 50) and adapter sequences [53,54]. Next, cp-like reads were extracted from trimmed reads using Basic Local Alignment Search Tool (BLAST) [55], with the cp genomes of S. miltiorrhiza (JX312195) being used as reference sequences. Finally, cp-like reads were used for genome assembly using SOAPdenovo [56]. Through a comparison of the contigs members and the lengths of different k-mer sizes (k-mer means that reads are divided into strings containing k bases and then assembled), k-mer sizes of 127 and 77, respectively, were found to provide the best results for S. przewalskii and S. bulleyana. The results from these two parameters were used to generate the final assembly. SSPACE and GapCloser were used to obtain scaffolds and fill gaps [56,57]. Cp genome annotation was performed using CPGAVAS with default parameters [58]. The result from CPGAVAS was corrected manually for start codons, stop codons, and intron/exon boundaries using Apollo [59]. The tRNA genes were identified using tRNAscanSE [60]. The circular maps of the two Salvia spp. were obtained using OGDRAW [61]. Codon usage and cp genomic characteristics were analyzed using MEGA6 [62]. The RNA editing sites of the gene-coding proteins in the two cp genomes were predicted using a predictive RNA editor, PREP suite [63], with a cut-off value set at 0.8.
《3.3. Characterization of repeat sequences and SSRs》
3.3. Characterization of repeat sequences and SSRs
Repeat sequences containing the three types of forward, reverse, and palindromic were identified using REPuter with a Hamming distance set at 3 and a minimum repeat size set at 30 bp [64]. Tandem repeats were analyzed using the Tandem Repeats Finder with default parameters [65]. SSRs were detected using MISA [66] with the following thresholds: eight repeat units for mono SSRs, four repeat units for di- and trinucleotide repeat SSRs, and three repeat units for tetra-, penta-, and hexanucleotide repeat SSRs [15].
《3.4. Genome comparison》
3.4. Genome comparison
The mVISTA [67] in the Shuffle-LAGAN mode was used to compare the cp genomes of S. przewalskii and S. bulleyana with the cp genomes of the other two reported Salvia spp. cp genomes (i.e., S. miltiorrhiza [JX312195] and S. japonica [NC_035233]) by using the annotation of S. przewalskii as the reference.
《3.5. Phylogenetic analysis》
3.5. Phylogenetic analysis
A total of 14 complete cp genome sequences were downloaded from the NCBI Organelle Genome and Nucleotide Resource Database for the phylogenetic analysis. The cp genomes were aligned using the MAFFT software, while MEGA6 [68] was used to construct the phylogenetic tree with the ML method. A bootstrap analysis was executed with 1000 replicates and tree bisection and reconnection (TBR) branch swapping, while Arabidopsis thaliana was set as the outgroup.
《4. Discussion》
4. Discussion
《4.1. Cp genome organizations》
4.1. Cp genome organizations
In this study, we are the first to report the complete cp genomes of S. przewalskii and S. bulleyana. The cp genome lengths of the two species range from 151 319 to 151 547 bp, with a typical quadripartite structure. These two cp genomes encode 114 unigenes, including 80 protein-coding genes, 30 tRNA genes, and four rRNA genes. Out of these genes, the CDS of the ycf2 gene is the largest, while that of the petN gene is the smallest. The 5' end of the rps12 gene is located in the LSC region, while its 3' end is located in the IR region, which is common in angiosperm cp genomes. In conclusion, the cp genome organization, GC content, gene number and type, and codon usage of S. przewalskii and S. bulleyana are similar to those of the two other Salvia cp genomes.
《4.2. Gene comparison》
4.2. Gene comparison
Some genes, such as matK–trnK, atpB–atpE, psbC–psbD, and rps3–rpl22, overlap each other in the eight Labiate cp genomes. A comparison of the overlap length shows that the overlap lengths of atpB–atpE, psbC–psbD, and rps3–rpl22 are the same, while the rps3 and rpl22 genes in Scutellaria baicalensis have no overlap. The length of the matK gene (1563 bp) is the same, but 12 variable sites were observed in S. przewalskii, S. bulleyana, and S. miltiorrhiza. The length of the matK gene (1530 bp) is the same in S. japonica, Mentha longifolia, and Scutellaria baicalensis. The matK gene was chosen by the Consortium for the Barcode of Life as the land plant barcode in 2009 [69]. A previous study has found that the matK gene can be used to identify different species in Labiatae better than the internal transcribed spacer (ITS) gene [70].
Here, we found two different ndhB gene types in the eight Labiate species; one encodes 511 amino acids, while the other encodes 493 amino acids. The length of exon 1 of the ndhB gene is 54 bp shorter in the two Salvia cp genomes than in S. miltiorrhiza (JX312195) when the length is annotated according to the cp genome of S. miltiorrhiza, where A substitutes for G in the eighth base position. This phenomenon causes a termination to appear, which is same as that in the ndhB gene in Perilla frutescens (NC_030756). This site was confirmed by polymerase chain reaction (PCR) with the forward primer AACAAACGAAAAGGAAACG and the reverse primer CTCCATAGGAACAATAGGG. The two exons of the fern ndhB gene have a unique pattern of intragenic copy number variants [71]. Although the rps16 gene is functionally lost in various legume lineages, reports show that the rps16 gene is essential even under heterotrophic conditions and can be functionally replaced by a nuclear gene [72,73]. In the present study, we found that this gene length is conserved in the four Salvia cp genomes, and only three variable sites were observed in this gene CDS (Table S8).
《4.3. Variance within genera》
4.3. Variance within genera
The technique of DNA barcoding, which was first proposed by Hebert et al. [74], can be used to identify species by means of DNA sequences such as ITS2, matK, psbA–trnH, and rbcL. However, the identification of proximal species—and particularly of morphologically confusing species in the same genus—still presents some difficulties. Therefore, finding a suitable DNA marker for such species is essential. The cp genomes have often been used for phylogenetic studies and species identification because of their slower evolution in comparison with nuclear genomes [75]. In the present study, an analysis of the pairwise cp genomic alignment and SNP in four Salvia cp genomes revealed an increased number of variable sites in the IGS of the trnV–ndhC, trnQ–rps16, atpI–atpH, psbA– ycf3, ycf1, rpoC2, ndhF, matK, rpoB, rpoA, and accD genes. Thus, these regions may be used as new candidate fragments to identify Salvia spp. In addition, ycf1a, or ycf1b is the most variable plastid genome region and can serve as a core barcode for land plants [76]. However, more Salvia cp sequence data support is required and should be addressed in future research.
《4.4. Relationship among the four Salvia spp.》
4.4. Relationship among the four Salvia spp.
S. przewalskii and S. bulleyana both belong to subgen. Salvia, while S. miltiorrhiza belongs to subgen. Sclarea and S. japonica belongs to subgen. Allagospadonopsis. This genetic relationship is reflected in the phylogenetic tree, where S. przewalskii and S. bulleyana are clustered together in one terminal branch. In terms of appearance and characteristics, many studies show that S. przewalskii and S. bulleyana are similar [77,78]. In terms of ingredients, S. przewalskii and S. bulleyana, which are often used as S. miltiorrhiza substitutes, contain most of the chemical content of S. miltiorrhiza; in contrast, S. japonica contains only some ingredients that are the same as those of S. miltiorrhiza [79,80]. In the phylogenetic tree, S. przewalskii and S. bulleyana form a robust monophyletic branch with S. miltiorrhiza first, and then with S. japonica. A total of 297 SNP sites were observed between the S. przewalskii and S. bulleyana genomes; this number is 785 among the S. przewalskii, S. bulleyana, and S. miltiorrhiza genomes. A total of 4982 variable sites were observed among the four Salvia cp genomes. All the analyses—including the repeat number and type, IR region borders, and phylogenetic tree—showed that S. przewalskii and S. bulleyana are closely related and that they differ more from S. japonica than from S. miltiorrhiza.
《5. Conclusion》
5. Conclusion
In this paper, we reported the complete cp genomes of S. przewalskii and S. bulleyana, which have long been used as S. miltiorrhiza surrogates in Southwest China. We also compared these two cp genomes with those of two other Salvia spp. published in the NCBI. The gene order and genome organization of the two Salvia species studied here are similar to those of the two Salvia cp genomes that have already been published. Moreover, repeated sequences containing SSRs were compared with six other cp genomes, revealing fine distinction. The variable site, IR region border, and phylogenetic analysis results showed that S. przewalskii and S. bulleyana are more similar to each other than to the two other compared species. The data and analysis in this paper may provide an in-depth understanding of the phylogenetic relationships between the species within the Salvia genus, and the complete cp genomes may be useful for future breeding and further biological discovery.
《Acknowledgements》
Acknowledgements
This work is supported by the National Nature Science Foundation of China (QFSL2018004, 2017YFC1702100, and 81741060) and the Fundamental Research Funds for the Central Public Welfare Research Institutes (ZXKT17004).
《Author contributions》
Author contributions
Shilin Chen and Jiang Xu conceived and designed the research framework; Baozhong Duan and Weisi Ma collected and identified the sample; Conglian Liang and Lei Wang performed the experiments; Conglian Liang, Xiaofeng Shen, and Shuai Guo analyzed the data; and Conglian Liang and Lei Wang wrote the paper. Shuiming Xiao, Haijun Qi, Zheng Wang, and Yaoqi Liu polished the language. Haoyu Hu, Juan Lei, and Jiang Xu made revisions to the final manuscript. All the authors have read and approved the final manuscript.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Conglian Liang, Lei Wang, Juan Lei, Baozhong Duan, Weisi Ma, Shuiming Xiao, Haijun Qi, Zhen Wang, Yaoqi Liu, Xiaofeng Shen, Shuai Guo, Haoyu Hu, Jiang Xu, and Shilin Chen declare that they have no conflict of interest or financial conflicts to disclose.
《Appendix A. Supplementary data》
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2019.01.017.











 京公网安备 11010502051620号
京公网安备 11010502051620号




