

《1. Introduction》
1. Introduction
Severe clinical infections caused by multidrug-resistant (MDR) Gram-negative bacteria (GNB) have become a critical threat to the public health worldwide [1]. Treatment strategies for infections caused by β-lactamase-producing GNB are limited, especially for those caused by carbapenem-resistant bacteria. Therapy with ‘‘last-line” agents (e.g., polymyxins and tigecycline) may be compromised by resistance, suboptimal pharmacokinetics, and/or high toxicity rates [2–4]. In February 2015, the US Food and Drug Administration (FDA) approval of a novel cephalosporin/β-lactamase inhibitor complex, ceftazidime–avibactam (CAZ–AVI), largely alleviated many of the concerns regarding traditional treatment options for MDR GNB infections [5–7].
Avibactam (formerly known as AVE1330A and NXL104) is a member of a class of β-lactamase inhibitors called diazabicy-clooctanes (DBOs) [8]. It has the capacity to rapidly acylate a wide range of β-lactamases while minimizing the liability of hydrolysis. CAZ–AVI thus exhibits activity against various clinically important β-lactam-resistant bacteria producing class A (e.g., extended-spectrum β-lactamases (ESBLs) and Klebsiella pneumoniae (K. pneumoniae) carbapenemases (KPCs)), class C (e.g., AmpC β-lactamases), and certain class D (e.g., oxacillinase (OXA)-48) enzymes, but not against the metallo-β-lactamases (MBLs) of class B (e.g., New Delhi metallo-β-lactamase (NDM), Verona integron-encoded metallo-β-lactamase (VIM), and imipenemase (IMP)) [9].
However, bacterial resistance is a potential risk from antibacterial usage. In the brief time since the introduction of CAZ–AVI in clinical use, pathogens developing resistance have been reported worldwide [10–14]. Therefore, there is an urgent need to understand the genetic basis for the emergence of CAZ–AVI resistance during treatment over a short timescale. In this work, we systematically review recent insights into the epidemiology, resistance mechanisms, and clinical use of CAZ–AVI, and discuss additional treatment options for CAZ–AVI-resistant GNB infections in order to provide possible clues for the development of novel strategies against the emerging problems.
《2. Characterization of avibactam and other β-lactamase inhibitors approved for clinical use》
2. Characterization of avibactam and other β-lactamase inhibitors approved for clinical use
At present, six β-lactamase inhibitors have been approved for clinical use. Sulbactam and tazobactam are penicillanic acid sulfones, and clavulanic acid is a clavam. All of these inhibitors function as ‘‘suicide” inactivators and take advantage of conserved active-site residues to interact with their targets, resulting in an irreversible inactivity of the targeted β-lactamase. The spectrum of these inhibitors is largely limited to some of the class A serine β-lactamase enzymes, such as temoneira (TEM)-1 [15]. The other three recently approved inhibitors are DBOs (avibactam and relebactam) [16,17] and boronic acid (vaborbactam) [18]. Unlike the ‘‘suicide” inactivators, they function as a competitive inhibitor by binding to targeted β-lactamases in a covalent but slowly reversible manner, followed by the regeneration of the active enzyme and intact inhibitor [16,19]. Although vaborbactam was initially designed to inhibit KPC-type carbapenemases, it also exhibits activity against other class A and class C β-lactamases [19]. However, meropenem–vabor-bactam shows less activity against strains that lack porins or that overexpress efflux pumps [18]. In 2019, the FDA approved relebactam in combination with imipenem and cilastatin. Imipenem–relebactam is active against carbapenem-resistant Enterobacterales (CRE) and carbapenem-resistant Pseudomonas aeruginosa (P. aeruginosa, CRPA), which produce KPC and class C β-lactamases. In some cases, imipenem–relebactam has shown good activity against carbapenem-resistant strains that lack porins, such as porin D (OprD)-deficient P. aeruginosa and outer membrane proteins OmpF/OmpK35 inactivate CRE [17,20]. Neither vaborbactam nor relebactam is able to inhibit class B and class D β-lactamases.
The addition of avibactam to ceftazidime can restore antibacterial activity against Enterobacterales and P. aeruginosa strains that produce a wide range of class A and class C β-lactamases [8,21]. Remarkably, avibactam is the only approved β-lactamase inhibitor that can assist ceftazidime to inhibit certain class D β-lactamases, such as OXA-48. In vitro studies have demonstrated that avibactam is highly efficient at inhibiting β-lactamases, such that only 1–5 molecules of avibactam are enough to inhibit one β-lactamase molecule, in comparison with 55–214 molecules of tazobactam and clavulanic acid [8]. It is notable that the reaction of KPC-2 with avibactam is irreversible, in that a β-lactam/β-lactamase inhibitor complex forms, which results in the hydrolysis of avibactam and regeneration of free KPC-2 [22]. In comparison with the other approved inhibitors, avibactam has the advantage of high efficiency in inhibiting β-lactamases, especially in the inhibition of OXA-48-type carbapenemase.
《3. Clinical use of CAZ–AVI》
3. Clinical use of CAZ–AVI
CAZ–AVI has been approved in the United States and Europe for the treatment of complicated urinary tract infections (cUTI), including pyelonephritis; complicated intra-abdominal infections (cIAI); and hospital-acquired pneumonia (HAP), including ventilator-associated pneumonia (VAP) [23,24]. In Europe, this combination is also approved for the treatment of infections caused by aerobic GNB in adult patients with limited treatment options [24]. Thus far, the drug has been approved in more than 40 countries and regions around the world, including China as of May 2019. This agent appears to be well tolerated in healthy subjects and hospitalized patients (pediatric and adult), with most adverse events being mild or moderate in intensity [25,26]. CAZ– AVI-based therapy has been reported to be associated with a significantly higher degree of both clinical success and survival rate in comparison with the other regimens employed for carbapenemresistant K. pneumoniae (CRKP) infections [5]. A study showed that the hospital mortality 30 days after patients were treated with either CAZ–AVI or colistin for CRE infections was 9% versus 32%, respectively [6], indicating a uniform superiority of CAZ–AVI over colistin. In addition, the rate of acute kidney injury has been found to be lower in patients receiving CAZ–AVI than in those treated by aminoglycoside combinations or colistin combinations [5]. These clinical studies consistently support a role for CAZ–AVI as a potential alternative to some ‘‘last-line” agents in the treatment of infections caused by carbapenem-resistant bacteria. The use of CAZ–AVI has been evaluated in 21 clinical studies, including one phase IV, six phase III, five phase II, and nine phase I studies as of April 2020 (Table S1 in Appendix A).
《4. Resistance mechanisms of CAZ–AVI》
4. Resistance mechanisms of CAZ–AVI
《4.1. Resistance induced by CAZ–AVI exposure》
4.1. Resistance induced by CAZ–AVI exposure
The emergence of CAZ–AVI resistance has been reported in both in vitro and in clinical practice. Livermore et al. [27] conducted an in vitro study to predict resistance-associated mutations using CAZ–AVI-susceptible KPC-3-producing Enterobacter cloacae (E. cloacae) and K. pneumoniae clinical strains. They revealed that CAZ–AVI selected mutants at up to 16× minimal inhibitory concentration (MIC), with frequencies of around 1 × 10–9. The first clinical case of CAZ–AVI resistance was detected in a KPCproducing K. pneumoniae (KPC-Kp) strain obtained from a patient with no history of CAZ–AVI therapy prior to the availability of CAZ–AVI [10]; treatment-emergent CAZ–AVI resistance has subsequently been reported in multiple centers worldwide [11–14]. The CAZ–AVI resistance mechanism is summarized in Table 1 [11– 13,28–44] and Table S2 in Appendix A. Current knowledge shows that the CAZ–AVI resistance mechanisms are complex and may be simultaneously mediated by multiple mechanisms in a single cell (Fig. 1). These are discussed below in detail.
《Table 1》
Table 1 Summary of CAZ–AVI resistance mechanisms developed in vivo.
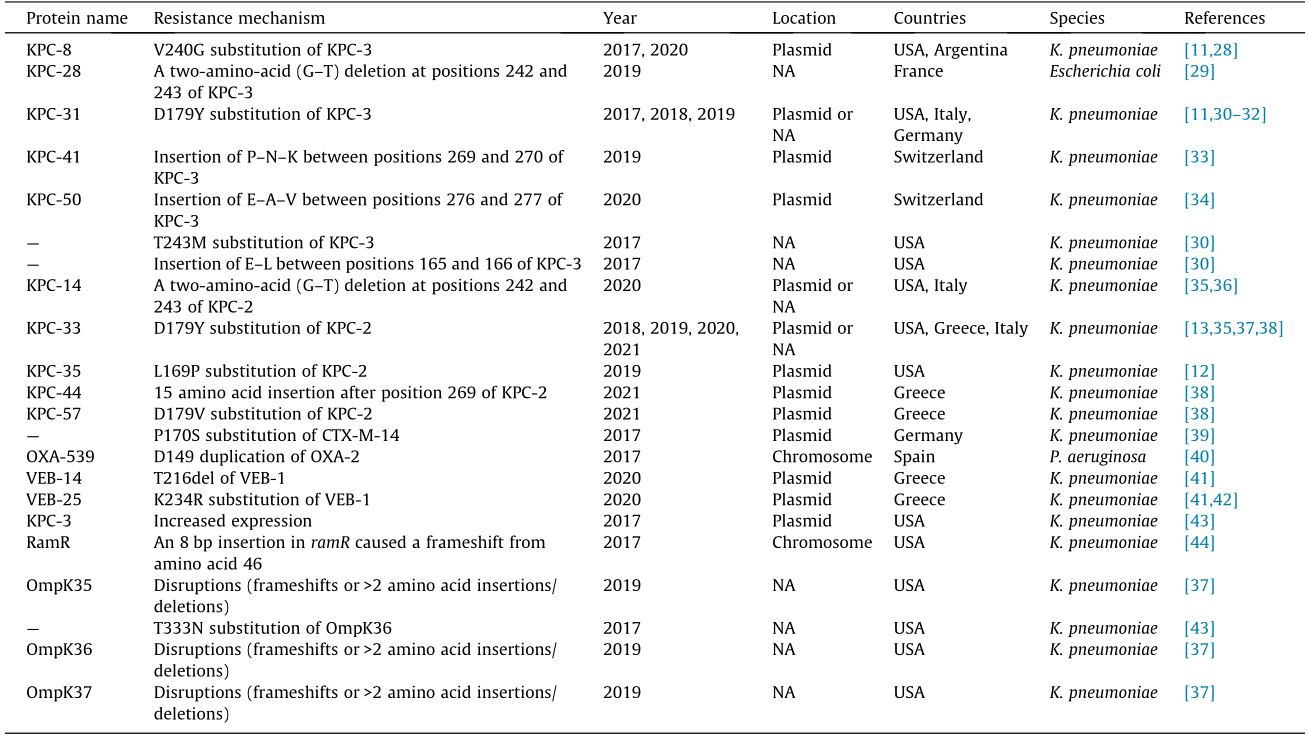
VEB: vietnamese extended-spectrum β-lactamase; G: glycine; T: threonine; P: proline; N: asparagine; K: lysine; E: glutamic acid; A: alanine; V: valine; L: leucine; bp: base pairs; NA: not available; CTX-M: cefotaximase; RamR: a multidrug-resistance regulator; —: the protein was unnamed.
《Fig. 1》

Fig. 1. A depiction of the bacterium with various resistance mechanisms of CAZ–AVI indicated. (I) Mutations occur in β-lactamases, including KPC, AmpC, CTX-M, and OXA-48; (II) production of metallo-β-lactamases that are unhindered by AVI; (III) overexpression of hydrolytic enzymes; (IV) enhanced efflux activity (AcrA/B–TolC and MexA/B–OprM); and (V) reduced cell permeability. AVI: avibactam; CAZ: ceftazidime; MBL: metallo-β-lactamase; PBP: penicillin-binding protein.
4.1.1. Mutations in blaKPC genes
To date, most cases of CAZ–AVI resistance have been caused by mutated blaKPC-2 and blaKPC-3 in K. pneumoniae (Fig. S1 in Appendix A). Structural studies revealed that these mutations frequently occur within the conserved motif region of class A β-lactamases named the Ω-loop, which encompasses amino acid residues Arg164 to Asp179 of KPC [45]. Remarkably, certain single amino acid substitutions within the Ω-loop, particularly at positions 164, 167, 169, and 179, can significantly reduce the susceptibility to CAZ–AVI [12,27,37,46]. Gaibani et al. [31] described the evolution of CAZ–AVI resistance by sequencing longitudinal clinical isolates from a patient with KPC-Kp bloodstream infection undergoing CAZ–AVI treatment. Using whole genome sequencing, a single amino acid substitution (D179Y) was found in KPC-3 produced by CAZ–AVI-resistant strains, when compared with the susceptible strain. The mutation of D179Y increases ceftazidime hydrolysis by creating a deeper pocket that traps the ceftazidime molecule for longer periods and avoids the binding of avibactam [47]. This dual effect exerted by the D179Y mutation thus largely enhances the resistance to CAZ–AVI. Another study showed that CAZ–AVI resistance was caused by the occurrence of a single mutation, L169P, in the KPC-2 enzyme produced by a K. pneumoniae strain. The strain was isolated from a patient who had accepted CAZ–AVI combination therapy with gentamicin for the treatment of VAP. Compared with the parental enzyme, the KPC-2 variant conferred an eight-fold increase to the MIC value of CAZ–AVI in Escherichia coli (E. coli) DH5a [12]. It is notable that CAZ–AVI resistance caused by Ω-loop substitutions can occur through the exertion of ceftazidime-related effects—that is, enhanced kinetics of ceftazidime—which are supposed to prevent the binding of avibactam. This mechanism has been confirmed in vitro [27,30,46] and in vivo [12].
Substitutions and deletions occurring in the Ω-loop and causing CAZ–AVI resistance have also been detected, such as V240G and T243M. Shields et al. [48] reported the development of CAZ-AVI resistance during the treatment of CRE infections for the first time since CAZ–AVI was approved for clinical use. CAZ–AVI-resistant K. pneumoniae emerged in three out of 37 patients after CAZ–AVI treatment courses, which was associated with treatment failure. An analysis of longitudinal CAZ–AVI-susceptible and -resistant isolates showed that the resistance was caused by amino acid substitutions in the KPC-3 enzyme (D179Y & T243M, D179Y, and V240G). Compared with the wild type, the three KPC-3 variants all increase the MIC values of CAZ–AVI ( ≥128-fold, ≥16-fold, and ≥4-fold) by being cloned into E. coli DH5α. The results suggest that, in ranking order, the impact of blaKPC-3 mutations on CAZ–AVI resistance is D179Y & T243M > D179Y > V240G [11]. However, the underlying resistance mechanism of substitutions in the Ω-loop remains unclear.
In addition to amino acid substitutions, amino acid insertions or deletions can confer CAZ–AVI resistance. Insertions of 1–15 amino acids in KPC-2 and/or KPC-3 have been reported to cause CAZ–AVI resistance (Table 1 and Table S2). For example, three KPC-2 mutants were identified in three CAZ–AVI-resistant K. pneumoniae strains isolated from three patients. Two of these strains harbored a D179Y and D179V substitution in the Ω-loop of KPC-2, respectively. A 15 amino acid insertion after position 259 was found in the third strain, and was designated as KPC-44 in that study [38]. A KPC-3 variant (designated as KPC-41), which obtained a three-amino-acid (P–N–K) insertion between position 269 and 270, was identified in a K. pneumoniae isolate, resulting in resistance to CAZ–AVI [33]. In addition, a variant of KPC-3 with a twoamino-acid deletion (△242-G–T-243), designated KPC-28, has been demonstrated to cause CAZ–AVI resistance in K. pneumoniae [29]. The same deletion is also found in a variant of KPC-2, KPC-14, resulting in a similar functional alteration for CAZ–AVI resistance in K. pneumoniae [36].
It is notable that mutated blaKPC genes conferring CAZ–AVI resistance can result in reduced or abolished carbapenemase activity [11–13,29,30,37,49] and become ESBL producers [30]. For example, the catalytic properties of KPC-2 harboring the D179Y substitution show impaired inhibition by avibactam with significant residual activity for ceftazidime hydrolysis [46]. However, this variant abolished the hydrolysis of aztreonam and imipenem.
4.1.2. Mutations in blaCTX-M genes
CTX-M-type enzymes are a group of class A ESBLs that have widely disseminated worldwide, and are originally inhibited by avibactam [50]. The acquisition of CAZ–AVI resistance due to mutations in the blaCTX-M-14 gene has been detected in a clinical K. pneumoniae isolate. Two nonsynonymous single-nucleotide polymorphisms were identified in the blaCTX-M-14 gene, resulting in two amino acid changes (P170S and T264I), one of which (P170S) was located within the Ω-loop. Compared with the blaCTX-M-14 wild type, the expression of the blaCTX-M-14 variants in the E. coli TOP10 strain showed a greater than 64-fold increase in ceftazidime MIC (from 4 to > 256 mg·L–1) and a 16-fold increase in CAZ–AVI MIC (from 0.5 to 8.0 mg·L–1) [39]. Another study reported that the simultaneous occurrence of two amino acid substitutions (L169Q and S130G) in CTX-M-15 caused CAZ–AVI resistance in vitro [51]. The dual substitutions resulted in the mutant being partially inhibited by avibactam at concentrations as high as 50 000 μmol·L–1, with neither of the substitutions being able to function independently [51]. Livermore et al. [52] identified one altered CTX-M-15 with a substitution of D182Y, which raised the CAZ–AVI MIC from 0.25 to 2 mg·L–1, but abrogated other cephalosporin resistance by plating ESBL producers on agar containing CAZ–AVI (1 or 4 mg·L–1). CAZ–AVI resistance mediated by mutations of CTX-M may have epidemiological significance in the future, as CTX-M is one of the most prevalent types of ESBL.
4.1.3. Mutations in blaVEB genes
Vietnamese extended-spectrum β-lactamases (VEBs) are a group of non-TEM, non-sulphydryl variable (SHV) ESBLs of Ambler class A. The residues comprising the avibactam binding pocket are known to be either identical or functionally conserved in various VEBs; thus, they can be inhibited by avibactam [53]. Most recently, mutations occurring in VEB-1 were found to cause CAZ–AVI resistance [53]. A novel variant of VEB-1, designated as VEB-25, was detected in two different KPC-Kp isolates resistant to CAZ–AVI. The isolates were obtained from two patients who had not received the drug in Greek hospitals in 2019. The avibactam was not able to inhibit the mutated VEB-1 enzyme due to a novel substitution, K234R [42]. Immediately following that publication, another group reported an outbreak caused by a CAZ–AVI-resistant K. pneumoniae strain coproducing KPC-2 and VEB-25 in a hospital in Athens, Greece. A total of seven patients were found to be colonized by CAZ–AVI-resistant K. pneumoniae strains, and three of them developed infections. The triple combination of CAZ–AVI + meropenem + fosfomycin or CAZ–AVI + aztreonam + fosfomycin was successful in the treatment of two of the cases at Day 14, while the combination of CAZ–AVI + meropenem was reported as a failure in the remaining case; unfortunately, all of the infected patients died by Day 28. This research group also reported a single hospitalized patient who was colonized by a CAZ–AVI-resistant KPC-Kp strain after receiving CAZ–AVI treatment one year before the outbreak. This strain was confirmed to produce another VEB-1 variant, VEB-14 (T216del, per Ambler numbering scheme), which exhibited decreased inactivation by avibactam [41]. The emergence of VEB-1 variants is a warning for us to maintain a sharp vigilance for occurrences of novel resistance mechanisms.
4.1.4. Hyperexpression and mutations of ampC genes
Alterations of Ambler class C β-lactamases are also involved in CAZ–AVI resistance. To understand the mechanism of resistance to CAZ–AVI and ceftolozane/tazobactam, Zamudio et al. [14] analyzed 24 P. aeruginosa isolates obtained from cystic fibrosis (CF) patients. They found that the resistance to ceftolozane/tazobactam and CAZ–AVI resulted from AmpC overexpression caused by 1,6- anhydro-N-acetylmuramyl-L-alanine amidase AmpD mutations. Likewise, elevated ampC gene expression was detected in six of nine CAZ–AVI-resistant P. aeruginosa strains isolated from adults with CF [54]. Alterations in the Ω-loop region of AmpC can induce CAZ–AVI resistance as well. For example, CAZ–AVI resistance was induced in three ceftazidime-resistant P. aeruginosa isolates in vitro, and various deletions (including five, seven, and 19 amino acid residues) were detected in the Ω-loop region of AmpC. The function of these mutations was demonstrated in the CAZ–AVI resistance [55]. Livermore et al. [52] consistently found that various AmpC modifications occurring in AmpC-derepressed Enterobacterales led to an increased MIC of CAZ–AVI, including substitutions of R168P/H, G176R/D, and N366Y, and deletions at positions 309–314. In another study, Compain et al. [56] cloned the N346Y variants of three highly divergent chromosomal and plasmid-borne AmpC β-lactamases to construct recombinant plasmids. After introducing each of the recombinant plasmids into E. coli TOP10, they found that the variants showed increased MICs of CAZ–AVI, and discovered that N346Y substitution was a likely route of acquisition of resistance to CAZ–AVI in AmpC β-lactamases. Given that some clinically important species of Enterobacterales, such as K. pneumoniae and E. cloacae, intrinsically carry ampC genes, it will be important to determine the occurrence of CAZ–AVI resistance caused by alterations of AmpC in these species in the future.
4.1.5. Mutations in blaOXA genes
At present, CAZ–AVI is the only approved β-lactamase inhibitor that is active against OXA-48; therefore, it has been used to treat infections caused by OXA-48-producing Enterobacterales [57]. However, CAZ–AVI resistance caused by OXA-48 mutations has been detected after exposure to CAZ–AVI in vitro. Double amino acid substitutions (P68A and Y211S) were induced in OXA-48, resulting in a five-fold reduction in the inhibitory activity of AVI [58]. The P68A substitution increases the flexibility and changes the plasticity of the substrate binding site, allowing the hydrolysis of bulkier drugs. The Y211S mutation affects the enzyme stability and confers higher ceftazidime resistance by altering the hydrogen bonding network. Together, the double substitutions reduce the inhibitory activity of AVI and specialize the carbapenemase toward ceftazidime hydrolysis [58]. Fortunately, no clinical OXA-48- producing isolates with CAZ–AVI resistance have been reported to date, which suggests that the mechanism mediated by OXA-48 mutations may be costly to bacteria.
It is notable that mutations in narrow-spectrum OXA β-lactamases can also cause CAZ–AVI resistance. In a clinical study, a CAZ–AVI-resistant P. aeruginosa strain was isolated from a patient with surgical infection. A three-base-pair insertion was identified in a blaOXA-2 gene, leading to the duplication of a key residue, D149. This duplication was demonstrated to be the determinant of CAZ–AVI resistance [40]. However, the underlying mechanism remains unknown.
4.1.6. Reduced permeability and overexpressed efflux pumps
As a ubiquitous strategy for drug resistance, porin mutations and efflux activity are involved in CAZ–AVI resistance as well [44,54,59,60]. Porin-deficiency, mutations, and downregulation are frequently identified in CAZ–AVI-resistant bacteria and synergize with other mechanisms, such as high ceftazidime hydrolysis activity [61] and KPC variants [49]. Although the entry of ceftazidime into the periplasmic space is thought to be less dependent on major porins (e.g., OmpK35 and OmpK36) than the entry of carbapenems [9], some clinical studies have claimed that major porins play a role in CAZ–AVI resistance. In an elegant study, two K. pneumoniae isolates were obtained from a single patient on two consecutive hospital days; one of the isolates was CAZ–AVI-susceptible and the other was CAZ–AVI-resistant. Both isolates encoded a nonfunctional OmpK35; the CAZ–AVI-resistant isolate additionally harbored a T333N substitution in OmpK36 and displayed higher expression of blaKPC-3 gene compared with the susceptible one [43]. The study demonstrated that the T333N substitution of OmpK36 decreased the susceptibility to CAZ–AVI; furthermore, a two-fold decrease of the CAZ–AVI MIC value was observed when the OmpK36 mutant was replaced by its wild type in the CAZ– AVI-resistant isolate. Likewise, the role of OmpK35 deactivity in CAZ–AVI resistance has been identified. A study showed that the expression of ompK35 decreased 28.5-fold in KPC-2-producing K.pneumoniae isolates with CAZ–AVI MIC greater than or equal to 1 mg·L–1 compared with those with MIC less than or equal to 0.5 mg·L–1. The ompK35 downregulation was caused either by frameshift or overexpressing of negative regulators. The susceptibility phenotype was able to be restored by a functional OmpK35, resulting in a two- to four-fold decrease in the MICs of CAZ–AVI [61]. Similar conclusions have been confirmed in other studies [54,59].
Efflux is not the primary pathway for CAZ–AVI resistance, yet the combination of enhanced efflux activity with other mechanisms associated with the resistance phenotype has been identified [44,60]. Nelson et al. [44] showed that mutations in the acrAB efflux operon regulator ramR result in the hyperexpression of the AcrAB–TolC efflux system in K. pneumoniae. With alterations of porins, such mutations contribute to CAZ–AVI resistance together. In addition, increased activity of the efflux pump MexAB–OprM and a high level of expression of ampC jointly promote CAZ–AVI resistance in P. aeruginosa isolated from CF patients[62]. The role of efflux pumps in CAZ–AVI resistance has been further demonstrated in a study that identified a variety of substitutions of OprD in ten CAZ–AVI-resistant clinical P. aeruginosa strains; the MIC value of CAZ–AVI dramatically decreased after the usage of efflux pump inhibitors. The study thus suggested that the major barrier for CAZ–AVI is membrane permeability and drug efflux [60]. Out of the nine CAZ–AVI-resistant P. aeruginosa isolates with elevated ampC gene expression described in Section 4.1.4, a loss of OprD was detected in seven strains, suggesting that porins and AmpC are co-involved in CAZ–AVI resistance [54]. Hence, the resistance mechanisms of CAZ–AVI among P. aeruginosa isolates are multifactorial, and may vary according to sample sources. It would be interesting to understand the fitness cost and prevalence of the various mechanisms involved in CAZ–AVI resistance, which could assist in determining empirical treatment with the rational use of CAZ–AVI in clinical settings.
《4.2. Production of β-lactamases unhindered by AVI》
4.2. Production of β-lactamases unhindered by AVI
The presence or acquisition of a β-lactamase (e.g., MBLs and most of class D enzymes) unhindered by AVI is a common mechanism of CAZ–AVI resistance [63,64]. MBLs hydrolyze most clinically available β-lactams, including carbapenems, and, thus far, cannot be inhibited by any of the commercially available β-lactamase inhibitors [65]. According to the zinc ion dependence and sequence similarity, MBLs are classified into three subclasses (B1, B2, and B3); the B1 subclass (i.e., NDMs, IMPs, and VIMs) is currently the most clinically important [65]. Class B1 MBLs are plasmid encoded and readily transferable among Enterobacterales with clinical significance, such as K. pneumoniae and E. coli [66], suggesting that the wide dissemination of MBLs presents a large challenge for the clinical use of CAZ–AVI. It is notable that, in contrast to the emergence of resistance described above, the production of β-lactamases unhindered by AVI can be regarded as baseline CAZ– AVI resistance.
《5. Global epidemiology of CAZ–AVI-resistant pathogens》
5. Global epidemiology of CAZ–AVI-resistant pathogens
The recent introduction of CAZ–AVI improves our ability to treat infections caused by MDR GNB, and especially those caused by CRE. However, with the increasing use of CAZ–AVI in clinical settings, the resistance rate is expected to continuously increase. As described above, it is of greater concern that CAZ–AVI resistance is mainly mediated by the mutations of resistance genes carried by self-transmissible plasmids, which greatly increases the widespread risk of CAZ–AVI resistance in the near future.
According to the available data, CAZ–AVI-resistant GNB mainly belong to Enterobacterales and P. aeruginosa. In a global surveillance program, the resistance rate of CAZ–AVI was evaluated for isolates recovered from respiratory and blood specimens collected from 879 patients with nosocomial-associated pneumonia. The resistance rate of Enterobacterales (n = 370) was 1.4% and that of P. aeruginosa (n = 129) was 11.6% [67]. In another global surveillance study (not including North America), Enterobacterales (n = 59 828) were found to have a resistance rate of less than 1.6% [68]. However, the resistance rate of CAZ–AVI seems to be geographically dependent. The data from Latin America showed that Enterobacterales (n = 7665) was highly susceptible to CAZ– AVI, with a low resistance rate of 0.3%, while the resistance rate of P. aeruginosa (n = 1794) was much higher, reaching 12.6% [69]. Results from the China Antimicrobial Surveillance Network in 2017 showed that the resistance rates were 5.4% and 13.5% for Enterobacterales (n = 1774) and P. aeruginosa (n = 524), respectively [70]. Taken together, all data consistently indicate that the CAZ–AVI resistance rate of Enterobacterales is much lower than that of P. aeruginosa. It is of greater concern that the P. aeruginosa isolated from CF patients showed a much higher resistance rate of CAZ–AVI, ranging from 24% to 57% [54,71,72]. We suppose that the higher resistance rate of CAZ–AVI observed in P. aeruginosa in comparison with that of Enterobacterales could be caused by various carbapenem-resistance mechanisms employed between them.
A recent epidemiological study tested 872 CRKP collected before the clinical use of CAZ–AVI in China, and showed that the resistance rate of CAZ–AVI was 3.7% [73]. Among the resistance isolates, more than half (53.1%) were MBL-producing K. pneumoniae, 40.6% were KPC-Kp, and the others (6.3%) were MBL and KPC co-producers. In another report of 232 CRKP isolates collected from a university hospital in China, the CAZ–AVI resistance rate was 8.2%. It was notable that nine of them were considered to be CAZ–AVI-resistant hypervirulent CRKP (hvCRKP) according to the results of a Galleria mellonella infection model and a mouse lethality assay [74]. Taken together, these data suggest that CAZ–AVI resistance in CRKP has emerged before the clinical use of CAZ– AVI, and that the newly emerged CAZ–AVI-resistant hvCRKP strains may represent another serious threat to the public health network.
《6. Clinical prospects》
6. Clinical prospects
At present, KPCs and OXAs are the major carbapenemases found in clinical settings, and are the targets of CAZ–AVI. We expect that the usage of this novel drug may change the epidemiological trends of carbapenemases, such as from KPCs to MBLs as major types [75]. In addition, the emergence of resistance with complicated mechanisms and an increasing trend caused by the horizontal transfer of self-transferable plasmids (e.g., KPCs are frequently plasmid borne) would alter the repertoire of drug-resistant bacteria in clinical settings [76]. Hence, active surveillance of the emergence of CAZ–AVI resistance is warranted. We advocate routine CAZ–AVI susceptibility testing for Enterobacterales, even in the absence of prior drug exposure.
An understanding of the local epidemiological patterns is important to guide the rational use of CAZ–AVI and to prevent the wide dissemination of CAZ–AVI resistance. It is also incumbent upon healthcare providers to share their clinical experience on the use of CAZ–AVI and other new β-lactamase inhibitors.
The reversion to carbapenem susceptibility that is caused by mutated blaKPC implies a possibility for carbapenems to be used to treat CAZ–AVI-resistant GNB [12]. Some have suggested the use of dual carbapenems plus CAZ–AVI therapy for CRE infections, which could theoretically attack both wild-type and mutated KPCproducing isolates [12]. Avibactam may protect carbapenems against hydrolysis by carbapenemases, and carbapenems can counter-select against blaKPC mutations that lead to CAZ–AVI resistance. However, the safety and efficacy of this combination in a clinical setting remains to be determined, and the stability of restored carbapenem susceptibility is transient. Furthermore, it is unknown whether other resistance mechanisms would be developed in this setting, which could result in treatment failure. It should be noted that other drugs may be used as salvage therapy once CAZ–AVI resistance is induced during treatments. For example, Athans et al. [37] reported a clinical case of meropenem–vaborbactam being used as a salvage therapy for CAZ–AVIresistant K. pneumoniae infection. Meropenem plus colistin treatment for 14 days was used successfully to treat recurrent pneumonia caused by CAZ–AVI-resistant K. pneumoniae [11]. In general, understanding the resistance mechanism of CAZ–AVI is crucial for making tailored strategies to effectively treat CAZ–AVIresistant bacteria infections.
Although the addition of avibactam greatly improves the activity of ceftazidime against most species of Enterobacterales, avibactam is unable to improve the activity of ceftazidime against Acinetobacter species or against most anaerobic bacteria (except for Prevotella spp. and Porphyromonas spp.) [77,78]. Currently, one of the greatest challenges in countering CRE is the development of inhibitors for MBLs. One promising product is aztreonam, which is a monobactam that is active against MBLs but inactive against isolates producing ESBLs, KPCs, and AmpC b-lactamases [79]. Therefore, the combination of avibactam plus aztreonam extends their potential utility against MBL-producing isolates that also encode serine β-lactamases. The activity of this combination against Enterobacterales coproducing MBLs and class A or class C β-lactamases has been demonstrated in a few studies [21,80]. For example, a study evaluated the activity of avibactam with ceftazidime, ceftaroline, or aztreonam against 57 well-characterized GNB and found that the aztreonam–avibactam combination was the only agent tested in the susceptible range (MIC, 0.12 mg·L–1) for VIM-1–TEM-1-producing E. cloacae isolates [80]. However, the aztreonam–avibactam combination would be inactive against the isolates coproducing MBLs and KPCs when variants are induced in KPCs by the aztreonam–avibactam treatment. Aztreonam– avibactam is currently in phase III clinical trials↑ . It remains to be determined whether other carbapenemase inhibitors currently in trials will be equally vulnerable to the rapid evolution of resistance and which genetic background may be particularly problematic.
↑ https://clinicaltrials.gov/ct2/show/record/NCT03329092
《7. Conclusion》
7. Conclusion
While the novel carbapenemase inhibitors currently fulfill an important need, they are unlikely to end the CRE epidemic due to the continuous emergence of complicated resistance mechanisms. The current status may be further fueled by polymyxin and carbapenem exposure. Our review highlights the need to optimize the clinical use of CAZ–AVI in order to minimize the emergence of resistance and to track the evolution of resistance in order to guide the development of novel treatments. A few outstanding questions still need to be answered in the near future: ① Would the clinical usage of CAZ–AVI change the epidemiological trends of CRE or the prevalent genotypes of carbapenemases? ② What is an effective salvage therapy for CAZ–AVI treatment failure? ③ How can we develop clinically safe and efficient inhibitors of MBLs (e.g., NDMs and IMPs) and other class D carbapenemases with clinical significance (e.g., OXA-23)? ④ How can we obtain the additional epidemiological data that is necessary to understand the prevalent resistance mechanism of CAZ–AVI in clinical settings?
《Acknowledgments》
Acknowledgments
This work was supported by the National Key Research and Development Program of China (2017YFC1200200), Major Infectious Diseases such as AIDS and Viral Hepatitis Prevention and Control Technology Major Projects (2018ZX10712-001), National Natural Science Foundation of China (81702045 and 81902030), and Shenzhen Basic Research projects (JCYJ20190807144409307).
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Tingting Xu, Yuqi Guo, Yang Ji, Baohong Wang, and Kai Zhou declare that they have no conflict of interest or financial conflicts to disclose.
《Appendix A. Supplementary data》
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2020.11.004.











 京公网安备 11010502051620号
京公网安备 11010502051620号




