

《1. Introduction》
1. Introduction
The human digestive system (DS) primarily consists of the mouth, esophagus, gastrointestinal tract, and accessory organs, such as the salivary glands, liver, and pancreas. DS cancers, which arise from the digestive tract and accessory organs, are currently the most common type of cancers. The most frequent DS cancers include esophagus, gastric, colorectal, liver, and pancreas cancers. The incidence of esophageal cancer, gastric cancer, and colorectal cancer (CRC) rank eighth, fifth, and third, respectively, among all malignancies worldwide [1]. Gastric cancer and CRC are also the third and second leading causes of cancer mortality worldwide [2]. Moreover, liver cancer and pancreatic cancer are the most lethal malignancies due to their late diagnosis and limited response to treatment [3,4]. Taken together, these cancers constitute more than 25% of cancer incidence and 35% of cancerrelated death in humans. Thus, it is an urgent clinical need to develop more effective treatment strategies for these cancers.
One of the major obstacles in the development of novel treatment is the lack of appropriate cancer models that can well recapitulate a patient’s tumors. In the past few decades, great efforts have been made to investigate the pathogenesis and treatment strategy of DS cancers with various models. At present, patientderived cancer cell lines (PDCs) and tumor xenografts (PDXs) are the most commonly used cancer models. However, two dimensional (2D)-cultured PDCs lack the features of the tumor architecture and microenvironment, which limits their applications in preclinical practice. PDXs have the advantage of mimicking the biological features of the human tumor better than PDCs. Nevertheless, they also have several drawbacks, including a low success rate and long propagation time.
During the last few decades, three-dimensional (3D) culture technologies have been used to develop novel and increasingly physiological human cancer models. It is worth mentioning that Sato et al. [5] successfully established 3D intestinal crypt-villus structures from a single leucine-rich repeat containing a G protein-coupled receptor 5 (LGR5+ ) stem cell, termed intestinal organoids. This study paved the way for researchers to establish organoids from other digestive organs. Thus far, organoid systems have already been established for most digestive organs, including the colon [6,7], liver [8], pancreas [9], stomach [10], salivary glands [11], and esophagus [12]. These 3D-cultured organoids, which recapitulate the histological features and preserve the genetic heterogeneity of the originating tissues, exhibit great advantages in comparison with PDCs and PDXs. Thus, organoids have become an ideal model in DS cancer research. In this review, we discuss the establishment methods and use of organoids in both basic and translational DS cancer research.
《2. Establishment of DS organoids》
2. Establishment of DS organoids
《2.1. Cell origins for generating organoids》
2.1. Cell origins for generating organoids
Theoretically, organoids can be generated from tissues or structures that contain stem cells. So far, the cell origins for generating normal organoids include pluripotent stem cells (PSCs) and adult stem cells (ASCs) (Fig. 1) [13–15]. ASCs exist widely in all tissue types and are responsible for tissue maintenance and injury repair. For example, studies have shown that LGR5 can be used as a stem cell marker in various kinds of digestive tract epithelium [16–18]. In the presence of multiple growth factors and other stimuli, tissue-derived ASCs can be embedded into a 3D matrix and grown into organoids with high efficiencies. Two kinds of PSCs—namely, embryonic stem cells (ESCs) and induced PSCs (iPSCs)—can be used to generate organoids. As human ESCs are neither convenient nor ethically permissible to use, they are not discussed here. Given their capacity for self-renewal and differentiation, iPSCs have been widely used in normal organoid constructions. However, the success rate of generating tumor organoids from patient-derived iPSCs varies among different cancer types and specific oncogenic mutations. Thus, iPSC-derived organoids, which are usually selected for the outgrowth of indicated tumor subclones, lack the genetic heterogeneity of the original tumors [19].
《Fig. 1》
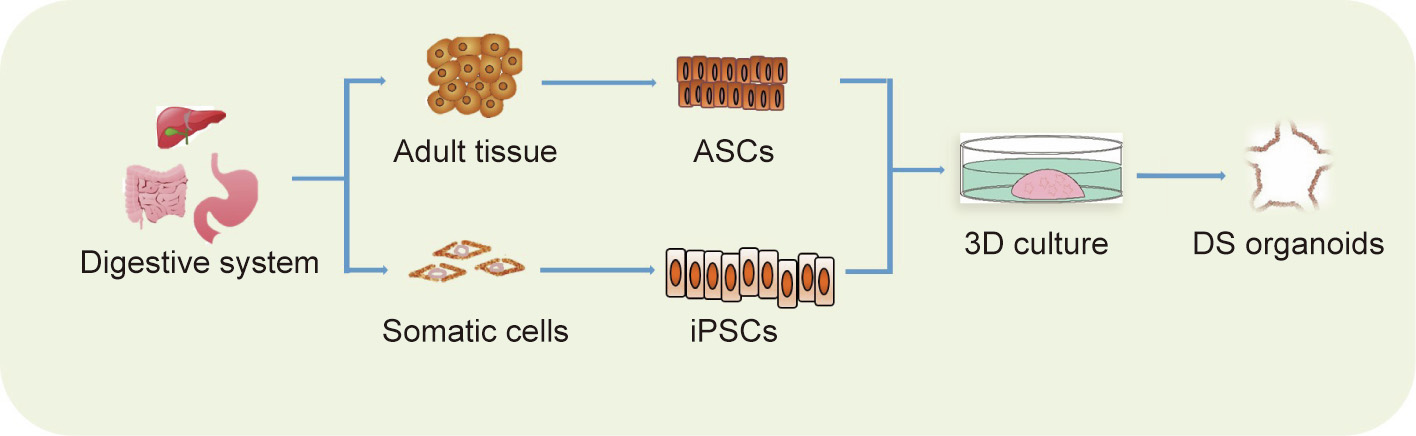
Fig. 1. The establishment of DS organoids. iPSCs: induced PSCs.
《2.2. General procedure》
2.2. General procedure
Numerous procedures have been reported for the construction of human DS organoids from mouse and human adult tissues [6,20–22]. Here, we describe a general procedure. First, redundant tissues such as muscular and connective tissues should be removed after the samples are collected. Then, the primary tissues should be minced into small pieces and subjected to tissue digestion in mild proteases such as collagenase or dispase. Single stem cells with specific stem cell markers can be sorted by flow cytometry. Next, digested cell mixtures or single stem cells are embedded in a 3D extracellular scaffold such as Matrigel, which assists in the differentiation and spatial organization of the cells into functional organoids [23]. A well-designed organoid culture medium containing a variety of growth factors, differentiation factors, and cytokines is added after the Matrigel solidification. The medium should be changed every 3–5 days, and single cells usually form 3D organoid structures within one week. These organoids can undergo continuous passaging and cryopreservation, while maintaining their morphology characteristics and genome stability.
The protocol of generating organoids from PSCs closely mimics the embryonic development processes. The procedure usually includes three steps: germ layer induction, tissue-specific spheroid formation, and organoid specification. As the DS comprises endoderm-derived tissues, endoderm patterning is essential for the generation of DS organoids [24–28]. In this protocol, activin A and bone morphogenetic protein 4 (BMP4) are used to induce the definitive endoderm [29]; next, a group of growth factors are supplemented to trigger foregut (Wnt family member 3A (WNT3A), fibroblast growth factor 4 (FGF4), a BMP inhibitor (NOGGIN)) or mid/hindgut (WNT3A, FGF4) differentiation. In addition, retinoic acid should be added on the last day to trigger the posterior fate in the foregut endoderm. Floating 3D spheroids will be observed 2–4 days after these treatments. Finally, the spheroids should be resuspended and embed in Matrigel, overlaid with the corresponding organoid culture medium [26].
《2.3. Culture system for DS organoids》
2.3. Culture system for DS organoids
The organoid culture media and extracellular matrix (ECM) comprise the organoid culture system. The system mimics the in vivo microenvironment for stem cell self-renewal and supports the in vitro expansion and 3D organization of stem cells.
Appropriate culture media, which contain a variety of growth factors, differentiation stimuli, and small-molecule compounds, are essential for the successful generation of organoids. All organoid cultures require a basal medium, which generally contains advanced Dulbecco’s Modified Eagle’s Medium (DMEM)/F12, penicillin/streptomycin, 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethane sulfonic acid (HEPES), GlutaMAX, N2, B27, nicotinamide, N-acetylcysteine, and other growth factors and inhibitors. Although there are differences in the components of the stem cell niche among DS organs, a series of factors, including receptor tyrosine kinase ligands (epidermal growth factor (EGF) and FGF10), Wnt activators (WNT3A and R-spondin), NOGGIN, and a transforming growth factor (TGF)-β inhibitor, are indispensable for the culture of most DS organoids. In addition, other factors or inhibitors will be supplemented to favor the expansion of DS organoids contingent on the tissue origin (Table 1 [6–10,12]).
《Table 1》
Table 1 Culture media for DS organoids.

As Wnt signals play important roles in the maintenance of active stem cells in the digestive tract [30,31], WNT3A and R-spondin are included in all the culture media for DS organoids. BMP signaling has been shown to inhibit the self-renewal of intestinal stem cells by suppressing Wnt/β-catenin signaling, and antagonistic BMP proteins are required for the long-term maintenance of stem cells [32,33]. EGF, which is associated with the proliferation of epithelium, is another important constituent of the culture media. FGF10 has been shown to promote cell proliferation and inhibit differentiation during digestive tract development; thus, it is essential for the culture of stomach, liver, and pancreas organoids [34]. Gastrin has been demonstrated to promote gastric cellular proliferation, and it is required in most culture media for DS organoids [35]. In addition to these growth factors, a set of small-molecule compounds are needed for the organoid culture. The Rho-kinase inhibitor Y-27632, which suppresses anoikis in single stem cells, is required for the effective generation of liver organoids [6,36]. Nicotinamide, also known as vitamin B3, has been shown to inhibit the activity of sirtuins and promote the formation of human colonic organoids [7]. As TGF-β signaling is reported to inhibit proliferation and promote apoptosis and differentiation in colon epithelial cells [37,38], the TGF-β inhibitor A83-01 is added to the organoid culture medium to increase the construct efficiency. The p38 inhibitor SB202190 has been found to improve the plating efficiency and promote continuous passages of human colonic organoids [39]. Prostaglandin E2 (PGE2) is reported to prevent detachment-induced cell death and activate canonical Wnt signaling cascade, and has been found to be necessary for human DS stem cell propagation [40] .
Unlike a 2D culture system, organoid cultures require an ECM to assist in the formation of spatially arranged and 3D patterned organ-like structures. The ECM consists of a large amount of biochemically distinct components with different physical and biochemical properties, including glycoproteins, proteoglycans, and polysaccharides [41,42]. Matrigel, which is extracted from mouse sarcoma, is currently the most widely used ECM in organoid culture systems. Matrigel consists of a variety of ECM proteins, such as collagens, laminin, and heparan sulfate proteoglycans, thereby providing a physiological environment for in vitro cells [43] .
《2.4. Criteria for organoid identification》
2.4. Criteria for organoid identification
So far, there is no uniform standard for organoid identification. To summarize the existing literature, the retaining of the morphological and genetic features of the original tissues is regarded as a common standard for organoid identification. Firstly, organoids should recapitulate the histopathological signatures of the primary tumors, which can be identified by histological analysis, including hematoxylin—eosin (H&E) staining and immunological staining. For example, liver organoids can be identified by the hepatocyte markers albumin (ALB) and hepatocyte paraffin 1 (HepPar1), and cholangiocyte organoids can be identified by the cyto-keratin 19 (KRT19) biliary marker. Organoids should also maintain the genetic characteristics of the primary tissues, which can be analyzed by means of genomic and transcriptomic sequencing. In our opinion, function representation should also be included as a necessary standard to identify organoids in future studies. The functional recapitulation of real organs enables organoid applications in regenerative medicines. For example, in an exciting study, Sampaziotis et al. [44] generated cholangiocyte organoids that can be used to repair human biliary epithelium after transplantation.
《3. Establishment of DS cancer organoids》
3. Establishment of DS cancer organoids
The organoid culture method for healthy DS epithelium has been successfully used in cultures of organoids from patientderived tumor tissue. To date, patient-derived tumor organoids have been successfully generated from multiple DS organs, including the esophagus [45,46], stomach [10], small intestine [47], colon [48,49], liver [50], and pancreas [9,51]. In general, surgically resected tumor tissues or biopsies are used to establish tumor organoids. Most of the composition of the culture medium of tumor organoids is identical to that of organoids derived from corresponding healthy tissues. However, organoids derived from Table 1 Culture media for DS organoids. human DS cancer samples often grow more efficiently in the absence of indicated niche factors, depending on the mutational background and activated pathways of the samples.
Esophageal cancers can be divided into two main histological subtypes: esophageal adenocarcinoma (EAC) and esophageal squamous cell carcinoma (ESCC). Studies have successfully established esophageal tumoroids from both human EAC and ESCC tissues [45,46]. Li et al. [45] generated organoid cultures derived from EAC that recapitulated its morphology, transcriptomic landscape, and genomic signatures. By utilizing a culture medium for murine esophageal organoids, DeWard et al. [12] and Kijima et al. [46] established both tumor and nontumoral organoids from the mucosa biopsies of ESCC patients.
Numerous studies have found that normal organoids grow more successfully than tumor organoids under optimized culture conditions, which may be related to genomic instability and resulting apoptosis in tumor organoids [48,50]. Thus, a normal organoid medium, as described above, will be modified by adding or removing the factors, in order to promote the growth of tumoroids and prevent the growth of nontumoral contaminating organoids. In the establishment of gastric tumoroids, for example, WNT3A is not needed for the culture of cancer organoids carrying the adenomatous polyposis coli (APC) gene mutation that leads to abnormal activation of Wnt signaling, EGF is not needed for cancer organoids carrying the rat sarcoma viral oncogene homolog (RAS)/mitogenactivated protein kinase (MAPK) gene mutation, and A83-01 is not needed for those carrying loss-of-function mutations of TGF-β gene [6,10].
CRC is a heterogeneous tumor with varying genetic alterations, clinical presentation, and prognosis in patients. It has been reported that the most commonly mutated signaling pathways include the Wnt, RAS/MAPK, phosphoinositide 3 kinase (PI3K), TGF, and tumor protein p53 (TP53) signals [52]. Thus, researchers have exploited the differences in niche factor requirements to selectively expand colorectal tumor organoids in diverse subtypes. Fujii et al. [49] established a set of colorectal tumor organoid lines from patients with different histological subtypes and clinical stages. These organoid lines were identified by transcriptomic signatures and selected according to niche factor requirements. The researchers demonstrated that the organoids well recapitulated the histopathological grade and differentiation capacity of their original tumor tissues.
Organoids of primary liver cancers, which include hepatocellular carcinoma (HCC), cholangiocarcinoma (CC), and combined HCC/ CC (CHC), have been successfully generated using a specific isolation and expansion medium [50]. Notably, to avoid the outgrowth of nontumoral organoids, Broutier et al. [50] modified the classical culture procedure of liver organoids by ① prolonging the tissue digestion time; and ② changing the culture medium by removing R-spondin 1, NOGGIN, and WNT3A, but supplementing dexamethasone and Rho-kinase inhibitor. As mentioned above, organoids can be alternatively derived from iPSCs, and several studies have reported the establishment of liver tumor organoids from iPSCs. For example, Sun et al. [53] established liver organoids by using reprogrammed human hepatocytes and the inactivation of p53 and retinoblastoma (RB), and then modeled the liver cancer organoids by genetically overexpressing oncogene c-Myc and RAS.
Several groups have successfully established pancreatic tumor organoids [9,54,55]. Similar to liver tumoroids, pancreatic tumoroids can be derived from different sources. Surgically resected tumor tissues are the primary source of pancreatic tumoroids. Boj et al. [9] generated organoids from both normal and tumorous pancreas tissues of mouse and human. With an orthotopically transplanted experiment, the researchers demonstrated that the pancreatic tumoroids completely reproduced the tumor development spectrum by forming early-grade neoplasms that progress to locally invasive and metastatic carcinomas. Another laboratory reported the successful modeling of pancreatic cancer with progenitor organoids from iPSCs by expressing mutant V-Ki-ras2 Kirsten ratsarcoma viral oncogene homolog (KRAS) or TP53 in the progenitor organoids [51].
《4. Organoids in DS cancer modeling》
4. Organoids in DS cancer modeling
《4.1. Host–pathogen interactions》
4.1. Host–pathogen interactions
Unlike other malignant tumors, DS cancers are significantly associated with pathogenic infections; examples include Helicobacter pylori (H. pylori) in gastric cancer, Salmonella enterica in gallbladder carcinoma, and hepatitis virus in HCC [56]. However, the association between infectious agents and cancer progression is still not well understood. Organoids that contain all the cell types of the original tumors can be used to study these processes via infection or co-culture systems with particular pathogens [57]. A number of studies have shown that gastric organoids can be used as an efficient H. pylori infection model. For example, Bartfeld et al. [10] utilized microinjection to infect gastric organoids with H. pylori and discovered the upregulation of gastric cancerassociated genes after infection. Another study by McCracken et al. [26] demonstrated that H. pylori infection leads to robust activation of cellular mesenchymal–epithelial transition factor (c-Met) and epithelial cell proliferation. Scanu et al. [58] exploited cocultures of murine gallbladder organoids with Salmonella enterica to examine the effects of infectious pathogens on gallbladder carcinoma development. Their findings suggested that Salmonella enterica leads to growth factor-independent growth and malignant transformation by activating AKT (also known as protein kinase B, PKB) and MAPK signaling. Viral infectious organoid models can also be established to study the virus–tumor relationship. For example, using primary intestinal organoids, Yin et al. [59] modeled rotavirus infection and assessed anti-viral medications. Similarly, liver organoids could serve as a potential model to study the link between hepatitis infections and hepatitis virus-related HCC development.
《4.2. Organoids in DS cancer progression models》
4.2. Organoids in DS cancer progression models
Cancer initiation and progression usually results from the gradual accumulation of mutations of tumor-driving genes [60]. Thus, it is of great importance to decipher the mutational processes that drive cancer development. Nowadays, whole-genome sequencing has been used to identify numerous genetic mutations that are associated with the development of DS cancers [61]. However, for most of these mutations, it has not been determined whether or not they have any tumor-driving functions. Previous studies on carcinogenesis have mainly been based on cancer cell lines and animal models. It is very inefficient to establish cancer cell lines from primary tissue, however, and the established cultures cannot fully retain the genetic signatures of the original tumor [62,63]. Although mousemodels are widely used to generate oncogenic mutations, it is time- and resource-consuming to establish genetically modified mice, and the interspecies differences between humans and rodents often influence the authority of the experimental results.
Organoids can remain genetically and phenotypically stable over a long period of culture. CRISPR-Cas9, a novel genome-editing tool, offers a method to introduce DNA double-strand breaks at specific genomic loci, making gene modification more convenient and efficient than previous genome editors [64]. A combination of CRISPRCas9 and organoids potentially overrides the abovementioned limitations. Recently, cancer initiation and progression models were established from healthy human colonic organoids. By utilizing CRISPR-Cas9 technology, four of the most frequently mutated genes in CRC (APC, TP53, KRAS, and mothers against decapentaplegic homolog 4 gene (SMAD4)) were sequentially introduced into healthy colonic organoids. The mutant organoids were selected by adjusting the composition of growth factors in the medium. The researchers found that the loss of APC and TP53 is the main driver of chromosome instability and aneuploidy in CRC [65,66]. Similar pancreatic cancer progression models were generated in human pancreatic ductal organoids using CRISPR-Cas9-mediated genome editing [54,67]. In addition to ASC-derived organoids, iPSC-derived organoids have been used to model the initiation and progression of digestive cancers. As mentioned above, liver cancer initiation has been modeled by modulating oncogenes or tumor suppresser genes in iPSCderived liver organoids [53,68]. Taken together, genetically modified human organoids will serve as a feasible and high-throughput tool to evaluate the significance of the mutations found in genome-wide sequencing.
《5. Drug development and personalized cancer treatment》
5. Drug development and personalized cancer treatment
During the last few decades, numerous anticancer drugs have been discovered through screening on traditional cancer cell lines. However, a significant percent of these newly discovered drugs have failed to achieve the expected outcomes in clinical trials [69,70]. Patient-derived tumor organoids retaining the genetic, transcriptomic, and histologic signatures of original tumors may serve as an advanced model to identify and test novel anticancer drugs. The drug sensitivity of tumor organoids has been demonstrated to be well matched with cancer molecular subtypes. For example, van de Wetering et al. [48] utilized CRC organoids to investigate the effect of a group of compounds. They found that the organoids carrying TP53 mutants were resistant to nutlin-3a (an inhibitor of murine double minute 2 (MDM2)) and those with RAS mutants were insensitive to EGF receptor inhibition. In addition, organoids with ring finger protein 43 (RNF43, a negative regulator of the Wnt pathway) mutants were found to be hypersensitive to Wnt inhibitors. An increasing number of studies have indicated that organoids can precisely reflect a patient’s response to anticancer drugs. Recently, Vlachogiannis et al. [71] established patient-derived colorectal and gastroesophageal cancer organoids and tested whether these organoids could predict patient treatment response to anticancer agents. The authors reported a high predictive value (positive predictive value of 88% and negative predictive value of 100%) of the patient-derived organoids (PDOs) in forecasting the response to targeted agents or chemotherapy in patients. Similarly, two recent studies demonstrated the successful use of rectal cancer organoids to predict patient responses to chemoradiation therapy [72,73]. In summary, these studies strongly demonstrate that PDOs recapitulate patient responses in clinical trials, laying a foundation for a new pattern of personalized medicine programs (Fig. 2).
《Fig. 2》

Fig. 2. The establishment and application of DS cancer organoids.
Aside from the efficient patient-response prediction of PDOs, another major advantage of employing organoids for drug development is the feasibility and economy of maintaining and amplifying them, which enable high-throughput drug screenings that are specific to individual patients [74]. Living cancer organoid biobanks can be used to perform drug screening and facilitate drug development. Thus far, living organoid biobanks of multiple DS tumor types including colorectal, pancreas, liver, and stomach, have been established [48–50,71,72,75,76]. For example, Yan et al. [76] established a human gastric cancer organoid biobank that includes diverse tumor subtypes. By testing the drug response to cisplatin and 5-fluorouracil (FU)-based drugs, the researchers found concordant responses of organoids with corresponding patients. Using the gastric cancer organoid biobank, the researchers performed large-scale drug screening and found new potential target drugs. In a study on biliary tract carcinoma (BTC), Saito et al. [77] established BTC-derived organoids and performed drug screening. They found that the antifungal drugs amorolfine and fenticonazole dramatically inhibited the growth of BTC organoids.
Moreover, intolerant side effects are one of the major obstacles that lead to drug failure in clinical trials. Thus, an excellent advantage of organoid technology is that both healthy and tumor organoids can be acquired from patients, which enables the screening of drugs that exclusively target tumor cells without harming healthy cells [78].
《6. Immunotherapy》
6. Immunotherapy
Although an increasing number of anticancer drugs have been uncovered, the development of drug resistance has resulted in limited therapeutic effect. In this context, immunotherapy, which involves stimulating the patient’s own immune system to combat malignant cells, is emerging as a powerful anticancer strategy. However, a lack of preclinical models to test the anticancer efficacy of such therapy has restricted its clinical applications. Increasing efforts are being made to integrate the immune system into organoid cultures. Several reports have shown that co-culture systems can be exploited to incorporate immune cells in organoid cultures. In a 2017 study using an air–liquid interphase method, researchers showed that CD45+ lymphocytes can remain alive for 8 days in coculture with human CRC organoids [79]. Using a similar method, Neal et al. [80] generated patient-derived cancer organoids that preserved the tumor microenvironment. In their study, immune cells including macrophages, T cells, B cells, natural killer (NK) cells, and natural killer T (NKT) cells successfully remained alive for 30 days in the co-culture system.
The success of organoid–immune cell co-culture systems provides an unprecedented model for anticancer immunotherapy research. In co-culture systems, the tumor cells in organoids can faithfully provide the antigen-specific stimulation of immune cells. Dijkstra et al. [81] utilized co-cultures of a patient’s own CRC organoids and peripheral blood lymphocytes to expand tumorreactive T cells. The obtained T cells were then used to assess the killing efficiency of matched lung cancer organoids. Chakrabarti et al. [82] tested the anticancer efficiency of programmed cell death ligand-1 (PD-L1) checkpoint inhibition via the co-culture of mouse-derived gastric cancer organoids with autologous immune cells. Chimeric antigen receptor (CAR)-based therapy is another essential immunotherapy strategy. In a 2019 study, Schnalzger et al. [83] tested the killing efficiency of CAR-engineered NK-92 cells using patient-derived CRC organoids. Therefore, based on the generation of organoids from various DS cancer types, co-culture systems of organoids and immune cells will serve as a valuable model to develop and evaluate immunotherapy in a personalized manner.
《7. Limitations and perspectives》
7. Limitations and perspectives
Despite the great potential of organoid technology as an outstanding in vitro cancer model, limitations remain. One of the most apparent disadvantages of organoid culture is the lack of cell type diversity, such as stroma, blood vessels, immune cells, and nervous cells, which prevents it from completely representing the complex tumor microenvironment. It is known that tumor microenvironments have a significant influence on tumor progression and drug resistance. Further studies should explore possible methods to incorporate additional cell types, such as the abovementioned coculture system. In a recent study, Workman et al. [84] recapitulated a normal intestinal enteric nervous system (ENS) by incorporating iPSC-derived neural crest cells into intestine organoids. The iPSC-derived nerve cells recombined with the organoids and successfully formed neuroglial structures similar to the ENS in the intestine, which showed neuronal activity. In addition, Öhlund et al. [85] were able to successfully establish co-cultures of murine pancreatic stellate cells with pancreatic ductal adenocarcinoma organoids. Moreover, the combination of 3D technology and organoid technology provides new opportunities for the use of organoids in studying the tumor microenvironment. Recently, Kim et al. [86] created multilayer bladder tumor ‘‘assembloids” by 3D-reconstituting tumor cells with stromal components to recapitulate the in vivo pathophysiological features of urothelial carcinoma. Another challenge is that cancer-tissue-derived organoids generally grow more slowly than normal tissue-derived organoids, resulting in the overgrowth of contaminating nontumoral organoids. Intensive explorations are needed to overcome this issue. As described above, Broutier et al. [50] successfully established liver cancer organoids of different subtypes by modifying the generation procedure and culture medium for normal liver organoids. It was recently reported that some CRC organoids grow more efficiently in medium without p38 inhibitor or under hypoxic conditions [49], indicating variations between normal and tumor organoids and even between tumor organoids.
At present, most organoid cultures require mouse-derived ECM substitutes (e.g., Matrigel or collagens) as scaffolds to support 3D cell growth. The ideal tissue scaffold analogue should not only consist of natural ECM proteins that can support the 3D growth of cells, but also be biocompatible and biodegradable. However, mouse-derived ECM, which contains complicated elements including laminin, collagen IV, and entactin, is not a well-defined matrix and may affect cellular activities and drug response [43]. Therefore, further investigations are needed to discover a more appropriate ECM analogue for organoid models.
In spite of these limitations, organoid technology has already developed into a new and promising in vitro model to facilitate the research of DS cancers. Thus far, both normal and tumor organoids can be efficiently generated from the DS tissues of patients. Applications of these organoids—especially PDOs—as preclinical models have been demonstrated to greatly improve cancer therapies and patients’ outcomes. In the future, the application of organoid technology will dramatically promote drug screening and personalized medicine, thus making the prediction of patientspecific drug responses and personalized therapy into a reality.
《Acknowledgments》
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81902904, 82073411, 81830054, and 81988101) and the China Postdoctoral Science Foundation (2019M661373).
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Xiaofang Zhao, Youhai Jiang, Chunliang Liu, Minghui Hou, Hongyang Wang, and Jing Fu declare that they have no conflict of interest or financial conflicts to disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




