

《1. Introduction》
1. Introduction
Metabolic diseases consist of a myriad of human maladies ranging from cancer to dysregulation in lipid metabolism. Disorders in lipid metabolism are the leading cause of obesity, type 2 diabetes mellitus (T2DM), non-alcoholic fatty liver disease (NAFLD), and cardiovascular disease (CVD). The etiology of these disorders converges on intracellular endoplasmic reticulum (ER) stress and inflammation induced by overnutrition and sedentary lifestyles. The functions of the ER are well established, including calcium regulation and lipid production, as well as protein synthesis, folding, and maturation [1].
Improper folding of de novo polypeptides often results in their accumulation in the ER lumen, leading to a phenomenon known as ER stress. As a compensatory mechanism, ER stress triggers several quality-control signaling cascades, including the activation of the unfolded protein response (UPR), autophagy, and ER-associated degradation (ERAD)mechanisms, in order to maintain ER homeostasis [2,3]. In mammalian cells, the UPR is mediated by three interrelated but independent transmembrane proteins: inositol-requiring enzyme 1α (IRE1α), activating transcription factor 6 (ATF6), and the RNA-dependent protein kinase (PKR)-like ER kinase (PERK). Under normal circumstances, these proteins are bound to several calcium-dependent chaperones, including the glucose-regulated proteins of 78 kDa (GRP78) and 94 kDa (GRP94). The accumulation of misfolded proteins in the ER causes a translocation of these chaperones from their cognate ligands to the unfolded proteins accumulating in the ER lumen. Release of these chaperones from the three transmembrane proteins results in the activation of several signaling cascades responsible for increased chaperone expression, protein degradation, and cellular apoptosis.
Chronic UPR activation and dysregulation of these pathways have been widely reported to contribute to an array of human diseases and metabolic disorders [4,5–14]. Chemical chaperones such as 4-phenylbutyric acid (4-PBA) have been posited to alleviate ERstress-driven metabolic disorders in mice and are under investigation in several clinical trials targeting obesity, diabetes, and other metabolic disorders [15,16]. Collectively, despite the established relationship between chronic UPR activation and disease progression, the distinct role of the ER-stress-induced gene, pleckstrin homology-like domain, family A, member 1 (PHLDA1) gene, in the context of a number of metabolic diseases has yet to be fully elucidated.
《2. Nomenclature》
2. Nomenclature
The murine homologue, T-cell death-associated 51 (TDAG51) gene, was initially identified based on its proapoptotic role in apoptosis antigen 1 (APO1/Fas)-mediated T-cell apoptosis [17]. Eventually, TDAG51 was classified as a PHLDA member, due to its high sequence homology with the PHLDA1 human homologue, with slight genetic variations encoding the N-terminal region of the protein [18]. Although TDAG51 has mainly been associated with cell death, the name PHLDA1 itself reinforces its multifaceted functional role, as does the other members of its family, which extends beyond apoptosis. In this review, the unified term PHLDA1 will be used exclusively, with the species defined by a lowercase prefix—namely, hPHLDA1 for the human homologue and mPHLDA1 for the mouse homologue.
《3. hPHLDA1 gene》
3. hPHLDA1 gene
The hPHLDA1 gene is located on chromosome 12q15, a region known to contain genes encoding ribosomal and nucleosomal proteins, along with a number of transcription factors [19]. The promoter region of the hPHLDA1 gene has been mapped to a 2210 nucleotide upstream region of the open reading frame, with the majority of promoter activity in the immediate proximal transcribed region being located 582 nucleotides upstream of the initiation start codon [20]. This nucleotide region demonstrates bidirectional activity, and the expression of the corresponding inverse transcript was identified in 293T cells through stimulation with ionomycin, an agent used to increase intracellular calcium levels, thereby triggering apoptosis and autophagy [20]. It has since been shown that expression of the hPHLDA1 gene can be subjugated by long non-coding RNA (lncRNA), microRNA (miR), and circular RNA (circRNA) interference. Recently, ENST00000552367, an 885 base pairs (bp) lncRNA transcript encoded by the antisense strand to PHLDA1 gene, was identified as a potential negative regulator of the PHLDA1 gene transcript [21]. The transcript of this lncRNA was 14-fold higher in gestational diabetes-induced macrosomia, resulting in a significant reduction in the PHLDA1 gene transcript [21]. A separate study identified another lncRNA having the ability to inhibit PHLDA1 gene, known as the hypoxia-inducible factor 1 α antisense RNA2. This inhibitory lncRNA is suggested to act as a scaffold for the lysine-specific demethylase 1-mediated epigenetic inhibition of PHLDA1 gene transcription in human trophoblast cells during the development of preeclampsia [22]. Another study revealed that PHLDA1 gene was a direct target gene of miR-101 [23]. While miR-101 mimics demonstrate significant repression of PHLDA1 gene, circ_0027599/miR-101 showed inhibition of miR-101, thereby restoring PHLDA1 expression [23]. Wang et al. [23] reported PHLDA1 regulation by circ_0027599/miR-101, which resulted in the suppression of gastric cancer survival and metastasis. While miR-194—also a PHLDA1 repressor—can be sequestered by lncRNA SNHGR1, this correlated with increased PHLDA1 expression in gliomas [24]. Additional research is required to determine the generalizability of the noncoding RNA regulation of PHLDA1 expression and its outcomes in metabolic diseases and cancer.
《4. Protein structure》
4. Protein structure
The full-length hPHLDA1 messenger RNA (mRNA) transcript is composed of 1955 nucleotides and contains two plausible start sequences [17,25]. Evidence suggests that the second internal start sequence is the major translation initiation site encoding a shorter 44 kDa hPHLDA1 protein [20,25]. The hPHLDA1 protein consists of 260 amino acids with the N-terminal half of PHLDA1 containing the structural PHLD region of the protein, resembling a unique split PHLD with seven β-sheets and a single α-helix interrupted between the third and fourth β-sheet by a 15 amino acid polyglutamine (poly Q) repeat (Figs. 1(a) and (b)) [26]. The PHLD region is followed by a proline–glutamine (PQ)-rich repeat region and a C-terminal proline–histidine (PH)-rich repeat (Figs. 1(a) and (b)) [17]. The mPHLDA1 protein contains 261 amino acids and shares 89.4% sequence conservation with hPHLDA1 (Fig. 1(c)). The expression of PQ/PH-rich proteins has been reportedly involved in transcriptional regulation and apoptosis in neuronal cells [25]. In Drosophilia, proteins with PQ/PH-rich repeats have been shown o have nucleotide-binding capabilities, suggesting that PHLDA1 may also have transcriptional activity, which is currently under investigation [27]. Moreover, overexpression of the PQ/PH-rich region of hPHLDA1 was found to be a potent inducer of cytotoxicity compared with full-length hPHLDA1 [28]. In contrast, the PHLD of PHLDA1 interacts with heat shock proteins to protect against cell death in human embryonic kidney (HEK293) cells and mouse embryonic fibroblasts (MEFs) [28]. The PQ regions introduce the possibility of several PHLDA1 isoforms with varying functionalities. The presence of PQ regions has been postulated to induce genomic instability, leading to altered protein function due to changes in protein length, as observed in several poly Qcontaining proteins [29]. The presence of PQ regions highlights the need for accurate reporting of PHLDA1 length when examining its expression in cancer and other metabolic disorders. Different isoform lengths of PHLDA1 could be investigated by designing functional assays to recognize specific segments of this protein. A deeper examination into the various isoforms and mutations of PHLDA1 gene could clarify the discrepancy in its pro- and antiapoptotic roles.
Classical pleckstrin homology domains are recognized for their ability to target proteins to the membrane bilayer containing phosphatidylinositol phosphate (PIP) and facilitate protein–protein interactions [26,30]. Pleckstrin homology domains are generally characterized by the two orthogonal β-sheets, each of which is composed of four β-strands, three variable loops, and a Cterminal α-helix, regardless of variations in the primary amino acid sequence [30]. Despite the split pleckstrin homology domains observed in PHLDA1, several biochemical studies have determined that proteins with split pleckstrin homology domains retain the conformation of the classical pleckstrin homology domain [31– 33]. In 1999, PHLDA1 was classified as a novel pleckstrin homology-related family member along with PHLDA2 (IPL/Tssc3) and PHLDA3 (Tih1), based on their predicted structural motifs and over 50% shared sequence identity (Fig. 1(d)) [18]. A comparison of functional roles among PHLDA family members has recently been reviewed, although PHLDA1 is disproportionately studied relative to the PHLDA2 and PHLDA3 family members [34]. One broad conclusion over the past decade is the functional redundancy among members, particularly involving the p53 and Akt serine/ threonine kinase 1 (AKT) pathways. Furthermore, both PHLDA1 and PHLDA3 genes are direct transcriptional targets of p53, while PHLDA2 has not been shown to be a direct target [35,36]. PHLDA1 and PHLDA3 also demonstrate the ability to bind to membrane PIPs through its PHLD, a vital interaction maintaining cytoskeletal organization [35,37,38].
《Fig. 1》
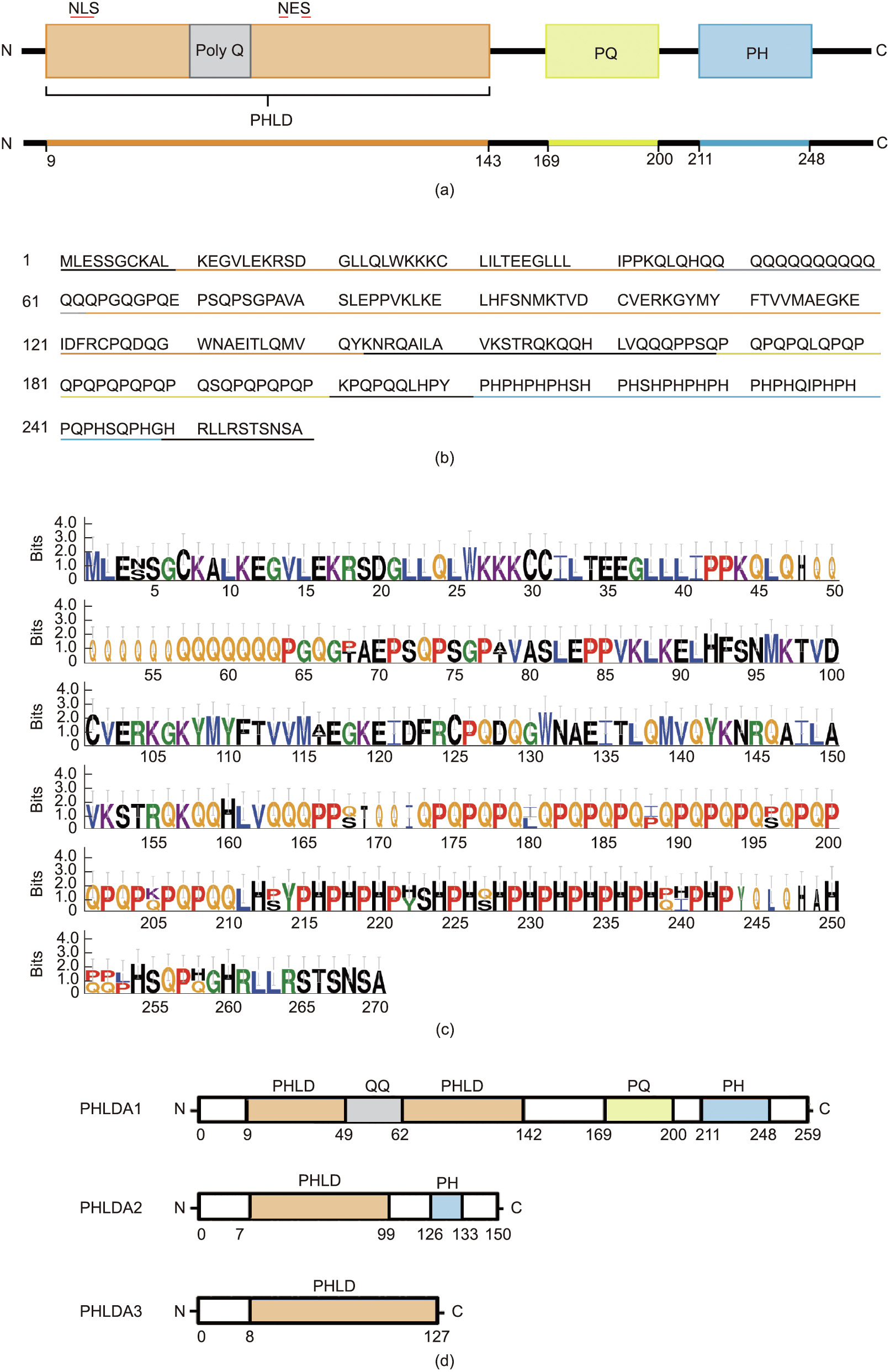
Fig. 1. PHLDA1 amino acid sequence and comparison with family members PHLDA2 and PHLDA3. (a) Protein schematic of hPHLDA1. hPHLDA1 consists of 260 amino acids with an N-terminal PHLD (orange) spanning 133 amino acids. PHLDA1 contains three additional regions of interest: a glutamine-rich linker (poly Q) (grey), a proline– glutamine (PQ)-rich repeat (yellow), and a C-terminal proline–histidine (PH)-rich region (blue). (b) Amino acid coding sequence of hPHLDA1, with corresponding regions underlined. (c) Sequence display similarity between human and mouse homologues of PHLDA1 generated using WebLogo 3.4 and Clustal multiple sequence alignment. The stack of letters symbolizes each position in the sequence. The overall height of each stack demonstrates the sequence conservation at that position (measured in bits), while the height of the letter reflects the relative frequency of the corresponding amino acid at that position. (d) PHLD family members. QQ: glutamin–glutamine.
To date, a crystal structure of PHLDA1 has yet to be resolved, with structural information primarily being based on sequence prediction alone. Using the Phyre2 protein-fold recognition server, the PHLD of PHLDA1 was mapped with 96.6% confidence to the split pleckstrin homology domain of the signaling protein, phosphatidylinositol 3-kinase (PI3K) enhancer (PIKE) (Fig. 2) [39]. PIKE is an integral Rho guanosine triphosphatase (GTPase) for PI3K signal transduction and has the ability to bind to PIPs through its split pleckstrin homology domain [40]. PIKE has several isoforms, including PIKE-L, PIKE-A, and PIKE-S; however, only PIKE-L and PIKE-S are known to predominately localize to the nucleus [40]. Furthermore, biochemical analysis of the split pleckstrin homology domain of PIKE-L revealed a functional nuclear localization sequence motif [41]. In contrast, a sequence analysis of PIKE-A identified a truncated nuclear localization signal and showed that it localized exclusively in the cytoplasm [41]. Given the high sequence homology between PIKE and PHLDA1, PIKE could be used to model and predict the functional role of PHLD in PHLDA1.
《Fig. 2》
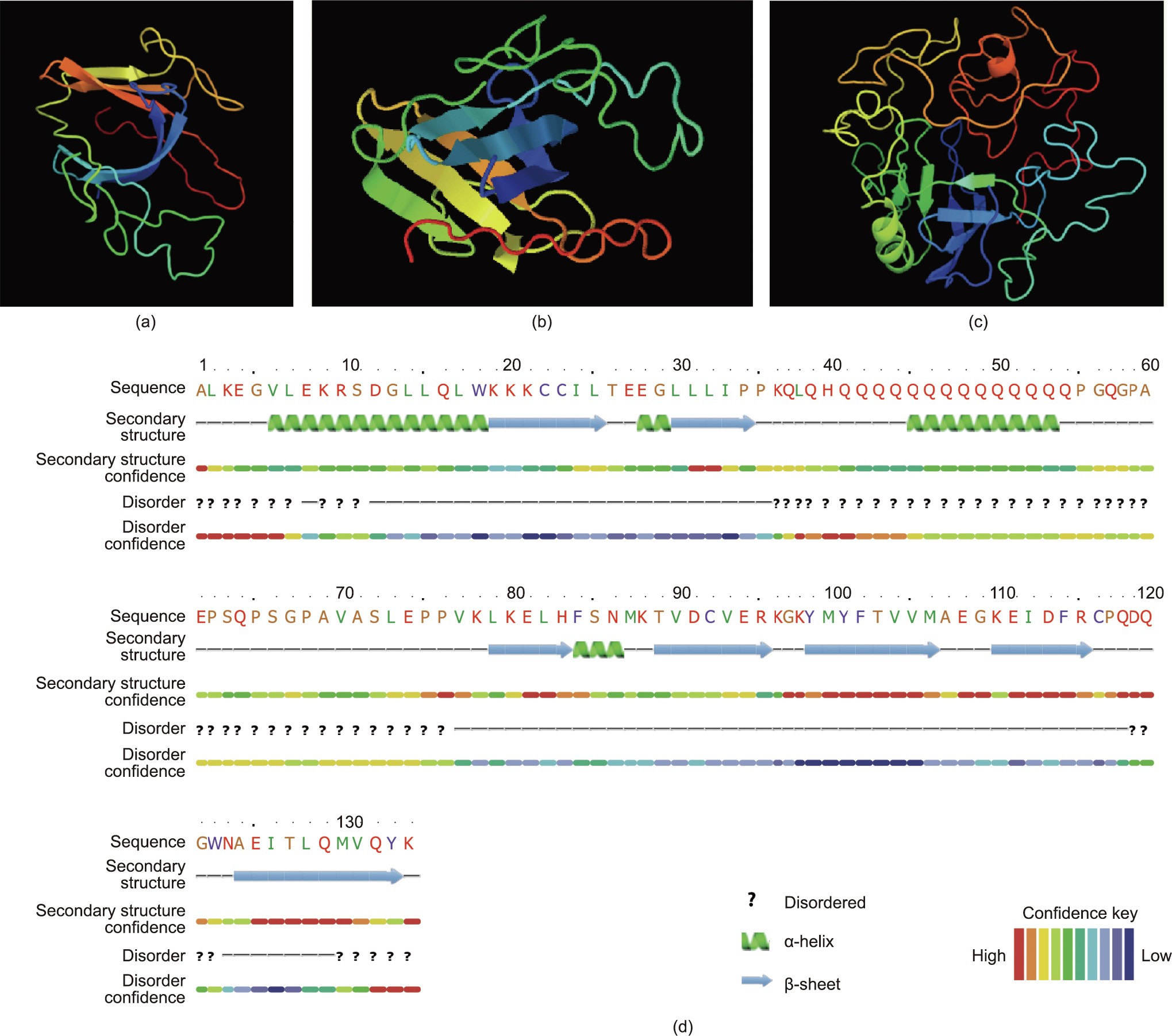
Fig. 2. Protein fold prediction models for PHLDA1 using Phyre2 (confidence interval is indicated using a color scale). (a, b) Three-dimensional (3D) model of the PHLD of PHLDA1 rendered using the split PH domain of PIKE and Phyre2 protein fold recognition server. Query of residues 9–141 of hPHLDA1 matched with 96.9% confidence to 86.0% of the sequence. Two alternative views of the model are shown. (c) 3D model of full-length PHLDA1, predicted using Phyre2. Large disordered regions loosely surround the structured β-sheet core. (d) Secondary structure and disorder prediction results for the PHLD of PHLDA1 from Phyre2. The seven β-sheets can be seen in the PHLD with a large N-terminal α-helix and secondary α-helix between the second and third α-helix, disrupting the seven-β-sheet organization of a classical pleckstrin homology domain. Reproduced from Ref. [39] with permission.
《5. Tissue expression and cellular localization》
5. Tissue expression and cellular localization
Basal PHLDA1 mRNA and protein expression have been documented in a wide range of mammalian tissues. Northern blot analysis of human tissue samples revealed high expression levels of PHLDA1 mRNA in the lung and pancreas, with moderate expression in the brain, heart, placenta, liver, and kidney [42–44]. Reportedly high levels of mPHLDA1 protein expression were observed in mouse lung and liver tissue, while being moderately expressed in the thymus and adipose tissue [45].
Altered PHLDA1 expression in cancer has been extensively reviewed [46]. In most cancers, the expression of PHLDA1 is reduced; however, this is not the case for colorectal cancer and osteosarcoma [34,46]. Chiu et al. [47] identified high expression of PHLDA1 in colorectal cancer—a direct contrast to hPHLDA1 expression in other cancers. PHLDA1 is normally expressed in cells within the crypt base throughout the intestine [48]. However, colorectal cancer tumors from mouse and human overexpress PHLDA1 mRNA relative to normal intestinal epithelium [49]. These results were further validated through immunohistochemical (IHC) staining of primary adenoma and carcinoma tumors in the small and large intestine [48]. Reduced expression of PHLDA1 in cancer cell lines suggests chemoresistance and varying susceptibility to cell death, which will further be discussed in this review [50].
Subcellular localization of PHLDA1 in human tissue varies widely and may affect the function of this protein. In human umbilical vein endothelial cells (HUVECs), PHLDA1 is localized to the cell periphery, with increasing perinuclear localization following homocysteine treatment [42]. Furthermore, the perinuclear localization of PHLDA1 is correlated with enhanced homocysteine-induced cell death [42]. The localization and function of PHLDA1 differ based on cell type, since strong cytoplasmic staining of PHLDA1 is observed in metastatic melanoma, which results in apoptotic resistance and growth deregulation [51]. Likewise, endogenous and forced expression of PHLDA1 predominantly localize in the cytoplasm and nucleoli of T-cells and result in the inhibition of protein synthesis [52]. A multitude of studies suggest that PHLDA1 localization may be associated with multiple functional roles in a tissue- and cell-dependent manner.
《6. PHLDA1 and its role in ER stress and cell survival》
6. PHLDA1 and its role in ER stress and cell survival
Expression of PHLDA1 gene is similar to that of other classical ER-stress-response genes [53,54]. Following exposure to a wide range of ER-stress-inducing agents, including thapsigargin, tunicamycin, farnesol, dithiothreitol, and cyclosporin, PHLDA1 expression levels are significantly upregulated, whereas agents that attenuate ER stress, such as salubrinal and 1,2-bis(o-aminophenoxy)ethane-N,N,N',N' -tetraacetic acid (BAPTA), mitigate PHLDA1 expression (Fig. 3) [42,55,56]. In addition, previous reports have shown that PHLDA1 is an inducible target of heat shock factor-1 (HSF-1) during proteotoxic stress [28]. HSF-1 has been shown to bind directly to the N-terminal PHLD of mPHLDA1 and substantially attenuate the apoptotic function of mPHLDA1 [28].
《Fig. 3》

Fig. 3. Agents that elicit ER stress or autophagy activate PHLDA1. Pointed arrows indicate activation, whereas flat discontinuous lines indicate inhibition. ER stress is generally activated by the accumulation of misfolded proteins. The ER-stress-inducing agents dithiothreitol (DTT) and homocysteine can cause the disruption of disulfide bonds (represented as S–S), leading to maladaptive proteins. Other ER-stress-inducing agents such as tunicamycin can cause misfolded protein aggregates by inhibiting Nglycosylation. ER stress can also be activated by inhibition of intracellular calcium transport: Thapsigargin inhibits the sarco-endoplasmic reticulum calcium adenosine triphosphosphatase (SERCA) pump, thereby directly depleting ER calcium and resulting in the ER stress activation of PHLDA1. The PERK pathway of the UPR is a compensatory response that is designed to restore proteostasis and regulate cell-death pathways. The PERK pathway can be activated by treatment with peroxynitrite, farnesol, and cyclosporin A. Downstream of the PERK pathway, an important regulator of cell death is eukaryotic translation initiation factor 2α (eIF2α), which can be attenuated with salubrinal. Inhibition of eIF2α has been shown to reduce PHLDA1 expression. High glucose, insulin, and insulin growth factor-1 (IGF-1) activate receptor tyrosine kinases, which in turn stimulate PI3K and PIP binding. PHLDA1 expression has been shown to be induced by PI3K and rapamycin-induced autophagy—a stimulus that may be repressed by AKT activity. HSF-1: heat shock factor-1; CHOP: C/EBP homologous protein, a potent inducer of cell death; Pi: phosphate.
The association between PHLDA1 and ER stress was characterized using a homocysteine-induced model [42]. Homocysteine treatment increased PHLDA1 expression and resulted in detachment-induced apoptosis or anoikis in cultured endothelial cells [42]. Treatment with tunicamycin—a N-glycosylation inhibitor and well-established ER-stress-inducing agent—in MEFs elevated PHLDA1 expression at both the mRNA and protein levels; however, cell death was not investigated as an outcome in this model [42].
Of the arms of the UPR, the PERK pathway is well established to promote the phosphorylation of the eukaryotic translation initiation factor 2α (eIF2α), which attenuates de novo protein synthesis [57–62]. In MEFs, phosphorylation of eIF2α was shown to promote mPHLDA1 expression, as mutations at the phosphorylation site of eIF2α were found to be critical for mPHLDA1 expression [42,63]. In support of these findings, salubrinal, a selective eIF2⍺ inhibitor, yielded a significant downregulation of hPHLDA1 in HK-2 renal proximal tubular cell lines after 18 h [55]. Moreover, hPHLDA1 is an important factor in the PERK pathway, since cytoplasmic PHLDA1 localization has been shown to strongly inhibit protein translation in 293T cells [52]. Overexpression of PHLDA1 was also shown to co-localize with a downstream effector of the PERK pathway: CHOP, a potent inducer of cell death [64]. Loss of PHLDA1 in mice protected against tunicamycin-induced tubular injury by impairing CHOP-mediated damage [64]. The cumulative evidence further reinforces the potential role of PHLDA1 as a downstream effector of PERK activation to modulate protein translation and cell death pathways in tandem with eIF2α and CHOP.
Although numerous studies have documented a relationship between PHLDA1 and apoptosis, the nature of this relationship is highly variable across different tissues and species. Upregulation of PHLDA1 is not universally proapoptotic, suggesting that this protein has a relatively complex homeostatic role. The proapoptotic properties of PHLDA1 were initially proposed to occur during Fas-mediated apoptosis [17]. Upregulation of PHLDA1 is correlated with activation-induced cell death (AICD), as initially demonstrated through a study on a T-cell receptor AICD-resistant T-cell hybridoma population [17]. This unique cell population was found to be deficient in PHLDA1 expression, implicating the protein in the T-cell receptor-mediated cell-death pathway and promoting its categorization as a proapoptotic protein [17]. Later, mPHLDA1 was found to be a non-essential component of Fas-mediated T-cell apoptosis in an in vivo study using mPHLDA1-deficient (mPHLDA1–/–) mice [65]. In the absence of mPHLDA1, no changes in T-cell AICD, the immune system, the physiology, or overall organ development were observed [65]. Another study further confirmed that hPHLDA1 expression is not correlated with AICD in human T-cells [66]. These conflicting results suggest a discrepancy between the initial in vitro study and the in vivo results with respect to PHLDA1’s role in AICD.
Early studies have shown a positive correlation between PHLDA1 expression and cell death in various cell lines, such as spermatocytes, MEFs, HeLa cells, and hippocampal cells [25,28,67]. It is notable that transient expression of PHLDA1 decreased cell survival in H19-7 hippocampal cells, providing evidence that PHLDA1 may mediate apoptotic processes. In another study, microinjection of H19-7 hippocampal cells with a neutralizing antibody specific to PHLDA1 increased overall cell survival and proliferation [25]. A positive correlation has been demonstrated between PHLDA1 expression in melanoma-derived cell lines and reduced cell growth and colony formation, as shown by an increase in the expression of cleaved caspase 9 and cleaved polyadenosine diphosphateribose polymerase activity (PARP) [51]. Similarly, transfection of PHLDA1 into HEK293 and Mel-Rif cells inhibited growth and colony formation by the induction of caspase-9- dependent apoptosis [51].
In contrast, the anti-apoptotic effects of mPHLDA1 in fibroblasts were demonstrated through an insulin growth factor-1 (IGF-1)- dependent mechanism. Small interfering RNA (siRNA) targeted against mPHLDA1 diminished IGF-1’s ability to rescue fibroblasts from serum starvation-induced apoptosis [68]. In embryonic fibroblasts, mPHLDA1 was shown to protect against reactive oxygen species-related stress [69]. In addition, PHLDA1 knockdown by siRNA in Ca9-22 cells increased activated caspase 3 expression, suggesting that PHLDA1 plays a role in suppressing apoptosis [70]. In MEFs derived from conditional Ewing sarcoma (EWS)/Friend leukemia virus integration site 1 (FLI1) gene knock-in embryos, EWS/ FLI1 expression resulted in apoptosis [71]. However, in these cells, PHLDA1 gene was shown to be directly repressed by EWS/FLI1 gene through direct binding of the PHLDA1 gene promoter [72]. Table 1 [17,25,28,51,67–70,72–75] summarizes the pro- and antiapoptotic functions of PHLDA1 based on cell type and state.
A counterpart to apoptosis is the autophagy pathway, a catabolic process that allows the recycling of degraded cellular contents as a renewable energy source [76]. Dysregulation of autophagy has been extensively reviewed in a myriad of diseases [77]. Rapamycin, a well-known autophagy activator and mechanistic target of rapamycin (mTOR) inhibitor, was shown to upregulate PHLDA1 expression in T-47 mammary cells. In this model, silencing PHLDA1 gene significantly reduced rapamycin-induced autophagy and apoptosis [73]. Recently, silencing of PHLDA1 gene in neuroblastoma cells resulted in a significant reduction of autophagy transcription and protein expression, further supporting the role of PHLDA1 as an inducer of autophagy [74]. The evidence for hPHLDA1-mediated autophagy regulation is limited and further investigation is warranted. Altogether, the current evidence suggests that PHLDA1 can promote autophagy. Limiting PHLDA1 to strict classifications as a pro- or anti-apoptotic protein fails to encompass its other physiological roles, such as its emerging associations with differentiation and regulating peroxisome proliferator-activated receptor 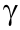 (PPAR) expression.
(PPAR) expression.
《Table 1》
Table 1 PHLDA1 expression associated with cell survival in multiple cell types.

HSP: heat shock protein.
《7. PHLDA1 mediation of adipocyte differentiation and white adipose tissue (WAT) expansion through negative inhibition of PPAR》
7. PHLDA1 mediation of adipocyte differentiation and white adipose tissue (WAT) expansion through negative inhibition of PPAR
Metabolic disease can manifest as a result of excess nutrient intake resulting in the pathological expansion of WAT through adipocyte hyperplasia and hypertrophy [45,78]. Previously, PHLDA1 was observed to be negatively associated with transcriptomic signatures of obesity in adipocytes [79]. Furthermore, inhibition of the neuropeptide Y-Y5 receptor in the hypothalamus of 12-week-old obese rats led to an upregulation of PHLDA1 in the adipose tissue and decreased body mass due to the reduction of both retroperitoneal and epididymal WAT, supporting the notion that PHLDA1 expression in adipose tissue protects against obesity [80].
In adipose tissue, PHLDA1 is also involved with a novel regulatory role in preadipocyte differentiation [45]. A previous study found that mPHLDA1 gene expression was upregulated during the early stages of differentiation in a microarray of 3T3-L1 preadipocytes [81]. During 3T3-L1 differentiation, mRNA expression of mPHLDA1 gene was enhanced within two hours of differentiation but was sharply attenuated by eight hours [45,81]. Concurrently, mPHLDA1 protein expression remained elevated for four days post-differentiation and was attenuated by day 14 [45]. Elevations in PHLDA1 gene transcript and PHLDA1 protein levels suggest a critical role in early preadipocyte cell conditioning. In support of this hypothesis, mPHLDA1–/– mice exhibited enhanced lipid accumulation and synthesis during adipogenesis, leading to a significant expansion in epididymal WAT despite similar food intake levels to wild-type controls [45]. mPHLDA1 shows an inverse correlation to PPAR during adipogenesis, with a downregulation of mPHLDA1 being associated with an upregulation of PPAR gene and increased adipocyte expansion [45]. In addition, both genetic knockout and siRNA silencing of mPHLDA1 gene lead to accelerated PPAR activation during preadipocyte differentiation, suggesting that PHLDA1 may be able to regulate adipose tissue expansion by inhibiting PPAR gene transcriptional activity [45]. Since then, mPHLDA1 has emerged as a novel negative regulator of PPAR. Forced expression of mPHLDA1 resulted in subsequent PPAR binding, which caused the inhibition of PPARc-driven-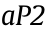 transcriptional activation and PPAR–retinoid X receptor α (RXRα) heterodimers in 3T3-L1 adipocytes [82]. PHLDA1 expression remains a key component of healthy preadipocyte differentiation and conditioning, while downregulation of PHLDA1 is potentially associated with excessive adipose expansion as observed in mPHLDA1–/– mice [45].
transcriptional activation and PPAR–retinoid X receptor α (RXRα) heterodimers in 3T3-L1 adipocytes [82]. PHLDA1 expression remains a key component of healthy preadipocyte differentiation and conditioning, while downregulation of PHLDA1 is potentially associated with excessive adipose expansion as observed in mPHLDA1–/– mice [45].
《8. Declining PHLDA1 levels associated with liver steatosis and injury》
8. Declining PHLDA1 levels associated with liver steatosis and injury
Obesity is characterized by chronic dysfunction in metabolic homeostasis and is often a precursor to other metabolic diseases such as glucose intolerance, insulin resistance, and increased system-wide lipid accumulation generally deposited in the liver and adipose depots. The most common comorbidities of obesity include T2DM, CVDs, and NAFLD [83,84]. It has been noted that varying models of obesity, including a high-fat diet, carbon tetrachloride, and leptin deficiency (ob/ob), all result in the direct loss of hepatic PHLDA1 protein expression [45]. More specifically, in ob/ob mice, detrimental depletion of hepatic mPHLDA1 mRNA and protein is associated with DNA hypermethylation [85]. In the same study, in vivo knockdown of mPHLDA1 gene by small hairpin RNA (shRNA) was found to increase hepatic lipid droplet size [85]. These findings further confirm the phenotype originally observed in mPHLDA1–/– mice, whereby loss of systemic mPHLDA1 expression results in significantly increased hepatic triglycerides through activation of sterol regulatory-element binding protein-1 (SREBP1)-target gene expression [45]. Following these findings, low levels of PHLDA1 were recently deemed to be a marker of nonalcoholic steatohepatitis (NASH) compared with normal healthy controls [86]. In contrast, PHLDA1’s role in maintaining liver homeostasis is strikingly opposite to that of its family member PHLDA3, which accelerates liver injury by promoting the IRE1–X-box binding protein-1 (XBP1) axis of ER stress [87]. Based on these findings, it may appear that PHLDA1 and PHLDA3 have opposing and possibly antagonistic roles in their response to liver injury; however, this direct interaction has yet to be shown.
As an extension of its homeostatic role in obesity and liver metabolism, loss of PHLDA1 results in insulin resistance and glucose intolerance in mice [45]. To date, the expression of PHLDA1 in diabetic patients has not been measured. However, a recent report identified the insulin-responsive phosphoenolpyruvate carboxykinase (PEPCK) binding site in the PHLDA1 regulatory sequence, classifying mPHLDA1 as an insulin-responsive gene [88]. The inhibitory effects of PHLDA1 on AKT signaling have been reported in the context of cancer; thus, it is important to investigate these interactions under the conditions of obesity and diabetes. Current evidence supports PHLDA1’s function as an essential regulator of adipose tissue mass, glucose homeostasis, and liver metabolism.
《9. PHLDA1 in atherosclerosis, inflammation, and vascular calcification》
9. PHLDA1 in atherosclerosis, inflammation, and vascular calcification
Atherosclerosis is a progressive disease characterized by the accumulation of plaque consisting of lipids, calcium deposits, and various fibrous elements, resulting in the narrowing of major arteries [89]. Immune cells and chronic inflammation are also heavily involved throughout the progression of atherosclerosis. Monocytes infiltrate from the bloodstream into the intima through the damaged endothelial cell wall, and subsequently differentiate into macrophages. The role of these macrophages primarily consists of metabolizing lipids through phagocytosis, transforming them into foam cells [90]. Oversaturation of low-density lipoprotein (LDL) and prolonged ER stress in these macrophages leads to apoptotic cell death, contributing to the formation of a necrotic core [91]. The release of proinflammatory cytokines and chemokines, along with vascular smooth muscle cell (VSMC) migration and extracellular matrix secretion, contribute to the formation of the fibrous cap that encapsulates the atheroma [92]. The presence of vascular calcification is a strong indicator of advanced atherosclerosis, whereby hydroxyapatite crystals form in the vessel walls, reducing arterial elasticity and altering atherosclerotic plaque stability [93,94]. Microcalcifications present in the fibrous cap of atherosclerotic lesions cause local stress that leads to cap rupture [95]. In contrast, larger calcium deposits provide moderate plaque stability and decrease stress on the fibrous cap by shouldering a portion of the mechanical load [96].
Emerging evidence suggests that PHLDA1 plays a crucial role in CVD (Fig. 4). In a case-control study of CVD, mutations in the hPHLDA1 gene were found to be significantly associated with CVD and myocardial infarction [54]. Downregulation of PHLDA1 was protective against atherosclerotic lesion progression through the modulation of apoptosis, cholesterol efflux, and peroxiredoxin-1 expression in mPHLDA1 and apolipoprotein E (ApoE)-deficient (mPHLDA1–/–/ApoE–/–) mice [54]. Loss of PHLDA1 gene in the same background of ApoE–/– mice was found to protect against atherosclerotic lesion growth and the development of necrotic areas by increasing cytoprotection against oxidative and ER stress relative to ApoE–/– mice [54]. Moreover, mPHLDA1–/–/ApoE–/– mice were observed to have enhanced PPAR-dependent reverse cholesterol transport in lesions (Fig. 4) [54]. Aortic roots from mPHLDA1–/–/ApoE–/– mice developed smaller atherosclerotic lesions, with a significant increase in the expression and nuclear localization of PPAR compared with their age-matched ApoE–/– controls [54]. PPAR has been found to play a role in the reverse transport of cholesterol in peritoneal macrophages, particularly those residing in atherosclerotic lesions [97]. Cultured mPHLDA1–/– peritoneal macrophages treated with LDL cholesterol (LDL-C) for 48 h displayed significantly less Oil Red O staining compared with wild-type cells, suggesting an enhanced underlying cholesterol efflux mechanism [54]. In line with these observations, isolated mPHLDA1–/– peritoneal macrophages treated with GW9662, a PPAR antagonist, exhibited similar levels of total cholesterol compared with wild-type peritoneal cells [54]. Taken together, these findings suggest that mPHLDA1 may also play a role in the facilitation and transport of cholesterol.
《Fig. 4》
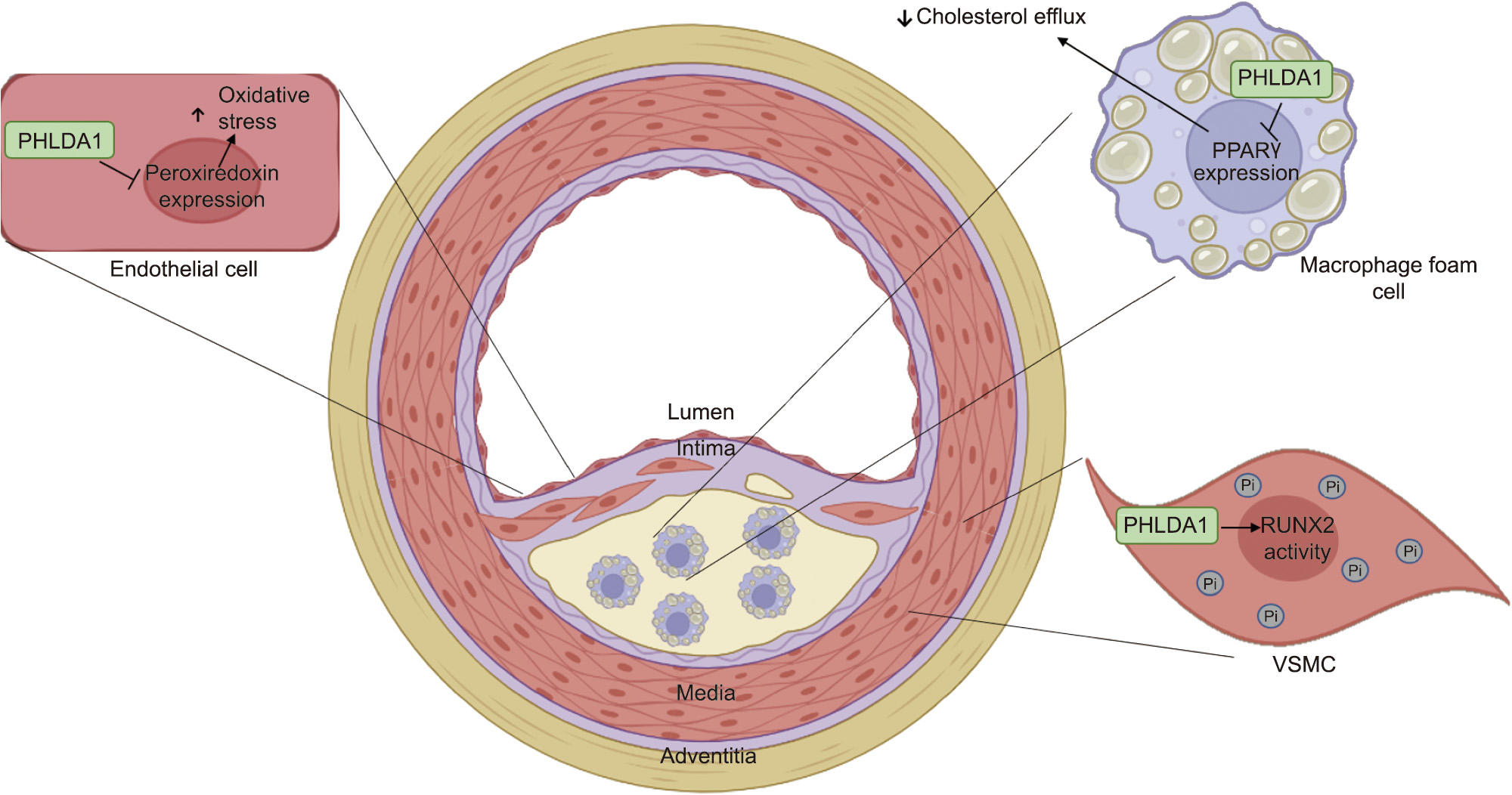
Fig. 4. The role of PHLDA1 in atherosclerotic lesion development: In endothelial cells, PHLDA1 inhibits the expression of peroxiredoxin and enhances oxidative stress, a known contributor to atherosclerotic lesion formation and progression. Elevated PHLDA1 expression also inhibits the PPAR expression, which reduces cholesterol efflux and enhances intracellular cholesterol accumulation in macrophage foam cells. In VSMCs, PHLDA1 is known to promote runt-related transcription factor 2 (RUNX2) gene transcriptional activity and the accumulation of intracellular Pi, as well as to contribute to vascular calcification.
Overexpression of hPHLDA1 has been shown to induce detachment-mediated programmed cell death or anoikis in human vascular endothelial cells [42]. Concurrent evidence also suggests a supporting role of mPHLDA1 in the progression of atherosclerosis under conditions of hyperhomocysteinemia. Aortic roots from ApoE–/– mice fed a high-homocysteine diet for a duration of four weeks showed prominent mPHLDA1 expression localized to necrotic areas within the atherosclerotic lesion [54]. Overall, these findings suggest a strong correlation between mPHLDA1 expression and the onset/progression of atherosclerosis through the upregulation of cell death and macrophage foam cell formation.
Lipopolysaccharide (LPS)-treated murine macrophage RAW264.7 cells were shown to have elevated mPHLDA1 expression [42]. Similarly, peritoneal macrophages isolated from mPHLDA1–/– mice elicit significantly lower LPS-induced macrophage chemoattractant protein-1 than wild-type controls [98]. In addition, in a lung-contusion mouse model, where lung contusion is a risk factor for acute lung injury and respiratory distress syndrome, downregulation of mPHLDA1 using siRNA decreased neutrophil infiltration and other inflammatory factors such as interleukin (IL)-1β, IL-6, tumor necrosis factor (TNF)-α, C–C motif ligand (CCL)-2, CCL-12, and Toll-like receptor (TLR)-2 [99].
In human and murine VSMCs, the absence of PHLDA1 has been shown to attenuate inorganic phosphate (Pi)-mediated hydroxyapatite mineral deposition in the medial layer of the vessel wall. Mechanistically, the loss of PHLDA1 expression reduced the cellular uptake of Pi, a main component of hydroxyapatite crystals, by reducing the expression of the primary Pi transporter found in the VSMCs, Pit-1 [100]. In the same study, mPHLDA1–/– mice were also found to be protected against medial vascular calcification in a vitamin D3 overload model. Mineral quantification and Alizarin Red staining revealed a decrease in mineral deposition in the aortas of mPHLDA1–/– mice, compared with wild-type controls. Primary mPHLDA1–/– VSMCs also displayed decreased expression of known drivers of vascular calcification, such as alkaline phosphatase (ALP), osterix (OSX), runt-related transcription factor 2 (RUNX2), and Msh homeobox 2 (MSX2) [100]. Overall, these findings suggest that PHLDA1 is a key player in the regulation of Pi-mediated medial vascular calcification.
《10. Conclusions》
10. Conclusions
n recent years, both homologues of PHLDA1 have been shown to play a critical role in a variety of pathophysiological conditions. The evidence outlined in this review demonstrates that PHLDA1 has both pro- and anti-apoptotic roles, which are dependent on cell type and disease state. Although both PHLDA1 mRNA and its translated protein can be regulated using various treatments and stimuli, the effects of this modulation have several implications related to cancer and lipid disorders, including the atherogenic progression, obesity, and fatty liver disease. Although PHLDA1, as a negative regulator of PPAR, is beneficial in the context of adiposity and fatty liver disease, this was found to be detrimental in atherosclerosis, where PPAR function is required for cholesterol transport. Future studies aimed at exploring the functional roles of PHLDA1 isoforms and mutations may reveal specific therapeutic targets. The literature should also focus on whether there is a compensatory effect of other PHLDA family members in the presence or absence of PHLDA1 expression and how this may contribute to disease progression. According to these important findings, PHLDA1 is a crucial regulator in several diseases; thus, further elucidating its role would allow for the development of novel therapeutic modalities targeting this underappreciated PHLD-containing cellular protein.
《Acknowledgments》
Acknowledgments
This work was supported in part by research grants to Richard C. Austin from the Heart and Stroke Foundation of Ontario (T-6146), the Heart and Stroke Foundation of Canada (G-13- 0003064 and G-15-0009389), and the Canadian Institutes of Health Research (74477). Financial support from St. Joseph’s Healthcare Hamilton is acknowledged. Richard C. Austin is a Career Investigator of the Heart and Stroke Foundation of Ontario and holds the Amgen Canada Research Chair in the Division of Nephrology at St. Joseph’s Healthcare and McMaster University.
《Authors’ contribution》
Authors’ contribution
Tamana Yousof and Jack Chen generated figures. Tamana Yousof, Jae Hyun Byun, and Jack Chen wrote the manuscript. Tamana Yousof, Jae Hyun Byun, Jack Chen, and Richard C. Austin revised the manuscript.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Tamana Yousof, Jae Hyun Byun, Jack Chen, and Richard C. Austin declare that they have no conflict of interest or financial conflicts to disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




