
《1. Introduction》
1. Introduction
Due to high patient compliance, cost-effectiveness, and fewer sterility constraints, oral drug delivery is the most preferred and convenient drug administration route [1–4]. Among such routes, there is a particular focus on intestinal-targeted drug delivery systems due to their unique advantages in the treatment of intestinal diseases and in the oral administration of protein and peptide drugs. Direct release of drugs in the pathology area can improve the local drug concentration, which is more effective for treating intestinal diseases, especially small intestinal tumors [5]. Some drugs, such as protein and peptide drugs, easily inactivate and degrade in the acidic stomach environment and thereby lose therapeutic effect. The bioavailability of these drugs can be significantly increased by intestinal-targeted drug delivery systems [6]. Moreover, certain drugs, such as indomethacin, have an irritating effect on the stomach and may cause stomach bleeding or perforation, which can be fatal [7]. Intestinal-targeted preparations ensure the direct delivery of drugs to the intestine, thus improving efficacy and reducing adverse reactions. In particular, intestinal-targeted drug delivery systems with controlled-release performance can achieve sustained release and long-term effects, which offer the advantages of enhancing drug safety and effectiveness, while reducing administration dose and frequency [2,8–13]. Most oral controlled-release drug delivery systems are designed as either a reservoir type [14–18] or a matrix type [7,9,10,12,19], based on a simple drug diffusion mechanism. With drug concentration difference as the driving force, constant drug release cannot be achieved with these systems, and the drug release can easily be affected by physiological factors in the gastrointestinal tract, leading to fluctuation of the drug concentration in the plasma. Furthermore, the above mentioned two systems are only suitable for the controlled release of water-soluble drugs. Therefore, the development of novel intestinal-targeted drug delivery systems to break through these limitations is of great significance.
Osmotic pump-based drug delivery systems driven by osmotic pressure difference can enable extended drug release characterized by desirable zero-order kinetics while hardly being affected by physiological factors [20–25]. The elementary osmotic pump (EOP) is mainly suitable for water-soluble drugs, and the push–pull osmotic pump was developed for water-insoluble drugs [22]. At present, most developed osmotic pump controlled-release preparations are osmotic pump tablets, which are composed of a tablet core containing drugs and osmotic active agents, a semipermeable coating film, and release holes on the film. Osmotic pump preparations using enteric polymers as the film-coating materials can realize intestinal-targeted drug delivery. For example, Chaudhary et al. [20] developed a bilayer-core osmotic tablet for colon-specific delivery, in which the colon-specific biodegradation of pectin in the coating film forms in situ pores for drug release. However, the coating process of these osmotic pump tablets involves the use of large amounts of organic solvents, and the preparation technology is complicated and costly [20–22]. Spherical capsules with sizes ranging from a few microns to a few millimeters show excellent application value as drug carriers to protect drugs from the surrounding environments [16–18,26– 30] and to release drugs in a controlled manner using appropriate capsule shell materials [9,14,15,18,19,31–33]. Compared with ordinary capsules, millimeter-sized capsules are more convenient for oral administration, as they can improve patient compliance; furthermore, compared with microcapsules, millimeter-sized capsules have a larger cavity structure that can encapsulate more drugs. Ca-alginate is an optimal material for constructing capsules for drug delivery due to its low cost, biocompatibility, non-toxicity, and simple fabrication process under mild conditions [9,10,16– 18,34–37]. In a previously reported work, we developed a pHresponsive Ca-alginate/protamine hybrid capsule featuring excellent protection of probiotics in gastric fluid and intestinaltargeted delivery characteristics [10,16,18]. However, the drug release still mainly depended on concentration-driven diffusion, so constant release was not achieved. Therefore, the development of Ca-alginate capsules with osmotic pump-based controlledrelease performance is highly desirable for intestinal-targeted drug delivery.
Here, we report on a novel θ-shaped Ca-alginate-based Janus capsule with double chambers for the intestinal-targeted constant release of drugs. As shown in Fig. 1, in the θ-shaped capsule, one compartment is designed as the ‘‘drug chamber” with its shell being embedded with enteric microspheres as ‘‘micro-valves,” while the other compartment is designed as the ‘‘booster chamber” and contains pH-responsive polymers as the pumping booster. Just like a microdevice with a push–pull osmotic pump effect, the proposed θ-shaped capsule can simultaneously achieve intestinaltargeted delivery and the controlled constant release of hydrophobic drugs through the intestine-specific dissolution of the enteric microspheres, which opens ‘‘microchannels” that release the drugs, and the intestine-specific swelling of the polymer booster, which provides a driving force for drug release. The double-chambered Ca-alginate-based capsules developed in this work hold high potential for intestinal-targeted delivery and the controlled pumping release of various active species.
《Fig. 1》
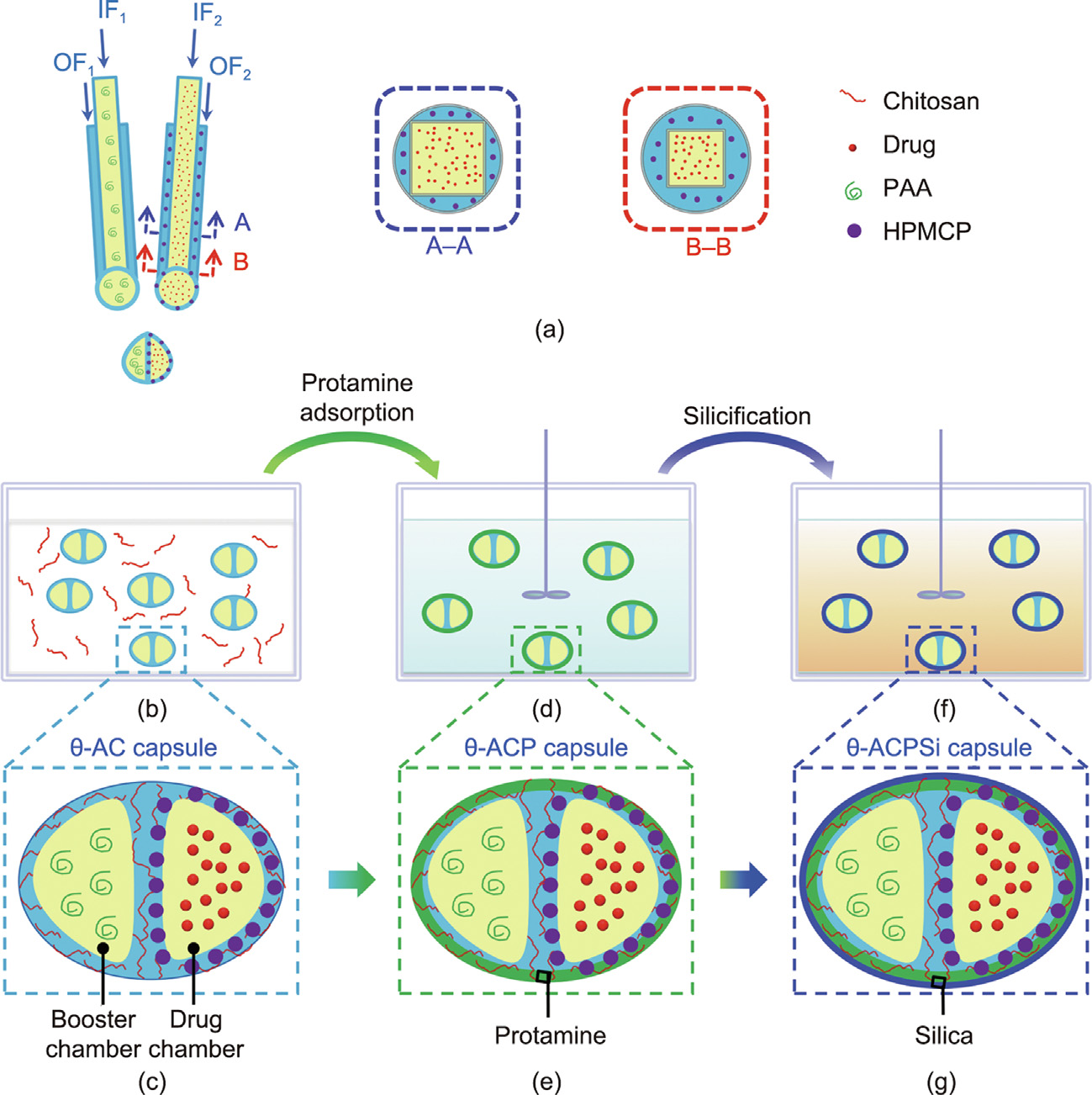
Fig. 1. Schematic illustration of the preparation process for the θ-shaped Ca-alginate–chitosan/protamine/silica (θ-ACPSi) capsules. (a) Composite co-extrusion minifluidic devices for fabricating Ca-alginate capsules with two chambers (booster chamber and drug chamber). (b, c) A θ-shaped Ca-alginate–chitosan (θ-AC) capsule with a booster chamber and a drug chamber, as well as hydroxypropyl methylcellulose phthalate (HPMCP) microspheres embedded in the shell of the drug chamber, prepared by the composite co-extrusion minifluidic devices. (d, e) A θ-shaped Ca-alginate–chitosan/protamine (θ-ACP) capsule prepared via an electrostatic adsorption approach. (f, g) A θ-ACPSi hybrid capsule prepared via a mild biosilicification process. IF1, IF2: inner fluid 1 and inner fluid 2; OF1, OF2: outer fluid 1 and outer fluid 2; PAA: polyacrylic acid.
《2. Materials and methods》
2. Materials and methods
《2.1. Materials》
2.1. Materials
Na-alginate, chitosan, and sodium carboxymethylcellulose (CMC-Na) were purchased from Chengdu Kelong Chemical Reagents (China). Hydroxypropyl methylcellulose phthalate (HPMCP; HP-55) was purchased from Shanghai Macklin Biochemical Co., Ltd. (China), polyacrylic acid (PAA; 40 kDa) was purchased from Hubei Baidu Chemistry (China), and indomethacin was purchased from Shanghai Aladdin Bio-Chem Technology Co., Ltd. (China). Rhodamine-labeled PAA (Rh-PAA; 40 kDa) was purchased from Xi’an Ruixi Biological Technology Co., Ltd. (China), and Lumogen Red 300 (LR300) was purchased from Tokyo Kasei Kogyo Co., Ltd. (Japan). Dulbecco’s modified Eagle medium (DMEM) culture medium with high-glucose penicillin–streptomycin solution, pancreatin, and fetal bovine serum (FBS) were purchased from Grand Island Biological Co. (USA). A Cell Counting Kit-8 (CCK-8) was supplied by Dojindo Laboratories (Japan). Pure water (18.2 MΩ at 25 °C) from a Milli-Q Plus water purification system (Millipore) was used throughout the experiments.
《2.2. Preparation of the h-ACPSi capsules》
2.2. Preparation of the h-ACPSi capsules
An assembly of two co-extrusion minifluidic capillary devices (Fig. 1) was used for fabricating the double-chambered capsules according to our previous work [35]. Typically, to form the booster chamber, an aqueous solution containing PAA and 1% (w/v) CMCNa was used as the inner fluid 1 (IF1), and 2% (w/v) Na-alginate solution was used as the outer fluid 1 (OF1). To construct the drug chamber, an aqueous solution containing indomethacin and 1% (w/ v) CMC-Na was used as the inner fluid 2 (IF2), while an aqueous solution containing 2% (w/v) Na-alginate, HPMCP microspheres, and 0.2% (w/v) surfactant sodium dodecyl sulfate (SDS) was used as the outer fluid 2 (OF2). CMC-Na was used to increase the viscosity of the inner fluids. The inner fluids and outer fluids were separately pumped into the inner square tubes and the outer cylindrical tubes by injection pumps, with the flow rates of IF1, IF2, OF1, and OF2 set at 25, 20, 10, and 15 mL·h–1 , respectively. At the nozzle ends of the two capillary devices, two droplets coalesced to form a single Janus θ-shaped droplet, which then fell into an aqueous solution containing 10.0% (w/v) Ca(NO3)2 and 0.2% (w/v) chitosan to form a θ-shaped Ca-alginate–chitosan (θ-AC) capsule at room temperature. The prepared θ-AC capsules were washed three times with 0.2 mol·L–1 acetic acid solution.
The θ-shaped Ca-alginate–chitosan/protamine/silica (θ-ACPSi) capsules were prepared using a biosilicification method [10,15,16]. In brief, the θ-AC capsules were immersed in 2 mg·mL–1 protamine solution for 30 min, allowing the protamine molecules to simply adsorb onto the surface of the θ-AC capsules. Then, these capsules were directly transferred into a 0.2 mol·L–1 acetic acid solution containing 60 mmol·L–1 sodium silicate for silicification for 1 h. Finally, the obtained θ-ACPSi hybrid capsules were washed three times with 0.2 mol·L–1 acetic acid solution and preserved in 0.2 mol·L–1 acetic acid solution.
《2.3. Investigating the ‘‘pumping effect” from the booster chamber》
2.3. Investigating the ‘‘pumping effect” from the booster chamber
The pumping ability of PAA was verified by comparing the swelling behaviors of alginate–chitosan (AC) and alginate–chitosan/ protamine/silica (ACPSi) capsules containing only the booster chamber under different pH conditions. According to our previous research, polymers with a small molecular weight may leak from the Ca-alginate-based capsules [15]. Therefore, PAA with an appropriate molecular weight (40 kDa) was chosen as the booster agent to prepare the Janus capsules. The PAA concentration does affect the drug release. Within a certain concentration range, the greater the concentration of PAA, the greater the osmotic pressure produced by PAA swelling, which is more conducive to drug release. However, the viscosity of the PAA solution increases with an increase in PAA concentration, which could affect the shear of the internal phase fluid and affect the construction of the double-chambered capsule structure [35]. Therefore, according to preliminary experiments, a PAA concentration of 0.5% (w/v) was chosen to prepare the Janus capsules with good morphology. The single-chambered capsules were fabricated using one coextrusion minifluidic device; the preparation parameters were similar to those of the double-chambered capsules, except that there was no drug chamber. The concentration of PAA in the inner fluid was 0.5% (w/v). These capsules were first immersed in a phosphate buffer solution (pH 2.5)-simulated gastric fluid for 3 h, and then transferred into a phosphate buffer solution (pH 6.8)- simulated intestinal fluid for 19 h. The swelling ratio Sw of the capsules was calculated by Eq. (1):

where V0 is the initial capsule volume in 0.2 mol·L–1 acetic acid solution, and Vi is the capsule volume at a certain time point.
《2.4. Investigating the pH-responsiveness of the HPMCP microspheres》
2.4. Investigating the pH-responsiveness of the HPMCP microspheres
Enteric HPMCP microspheres were prepared via microfluidic technology combined with a solvent diffusion method (Fig. S1 in Appendix A). The inner fluid was a mixed solution of dichloromethane and ethanol (9/1, v/v) containing 0.7% (w/v) HPMCP, the outer fluid was an aqueous solution containing 0.6% (w/v) SDS, and the receiving liquid was deionized water. The flow rates of the inner and outer fluids were 500 and 1400 μL·h–1 , respectively. Finally, the fabricated HPMCP microspheres were washed with pure water and then freeze-dried. To study their enteric properties, the freeze-dried HPMCP microspheres were dispersed in simulated gastric fluid and simulated intestinal fluid, respectively. The morphology and particle size of the HPMCP microspheres were observed using a microscope (SZX16, Olympus, Japan).
《2.5. Morphological characterizations of the capsules》
2.5. Morphological characterizations of the capsules
The morphologies of the θ-AC and θ-ACPSi capsules were characterized using a digital camera (E-PL5, Olympus, Japan) and a confocal laser scanning microscope (CLSM; SP5 II, Leica, Germany). To confirm the double-chambered structure, different fluorescent dyes were loaded into the distinct cores: Water-soluble Rh-PAA (instead of PAA) was added into IF1 to prepare the θ-AC capsules, and oil-soluble fluorescent LR300 (instead of indomethacin) was added into IF2 to prepare the θ-ACPSi capsules.
《2.6. Stability of the θ-ACPSi capsules in different buffer solutions》
2.6. Stability of the θ-ACPSi capsules in different buffer solutions
A swelling experiment was performed with the capsules for more than 20 h to explore the long-term stability of the θ-ACPSi capsules in the different pH environments of the gastrointestinal tract, so as to demonstrate that the whole capsule could maintain a state with almost no swelling for a long time at pH 6.8. The θ-ACPSi capsules were first immersed in simulated gastric fluid for 3 h, and then transferred into simulated intestinal fluid for 19 h. The long diameters and short diameters of the θ-ACPSi capsules were recorded by a digital camera at predetermined time intervals. The microstructures of the θ-ACPSi capsules at pH 2.5 and pH 6.8 were characterized by means of scanning electronic microscopy (SEM; G2 Pro, Phenom, China). A compressive test was performed on the θ-AC capsules and θ-ACPSi capsules before and after siliconization using an electronic universal testing machine (EZ-LX, Shimadzu, Japan).
The prepared drug-loaded θ-ACPSi capsules were randomly divided into four groups, each with ten capsules. One group was taken to measure the average value of the initial drug content of a single capsule, while the remaining three groups were placed in test tubes containing 0.2 mol·L–1 acetic acid solution and their drug release amounts were measured regularly at 0, 7, 14, 21, and 28 d. According to the initial drug content of a single capsule (m0) and its drug release amount at a certain time point (wi), the rate of change (Ri) of the drug content of the capsules at time point was calculated by Eq. (2):

《2.7. In vitro drug release behavior of the θ-ACPSi capsules》
2.7. In vitro drug release behavior of the θ-ACPSi capsules
Using indomethacin as the model drug, the drug release behavior of four kinds of drug-loaded θ-shaped capsules in simulated gastric fluid and simulated intestinal fluid was investigated. These four kinds of drug-loaded capsules were: θ-ACPSi capsules containing only HPMCP (HPMCP@θ-ACPSi); θ-ACPSi capsules containing only PAA (PAA@θ-ACPSi); θ-ACPSi capsules containing HPMCP and PAA ((HPMCP + PAA)@θ-ACPSi); and θ-ACPSi capsules without HPMCP or PAA. A certain number of capsules were immersed in a diffusion device containing simulated gastric fluid at 37 °C for 3 h. At predetermined time intervals, the concentration of indomethacin in the surrounding medium was analyzed by means of ultraviolet–visible spectrophotometry (UV-1800, Shimadzu) at a wavelength of 320 nm. The capsules were then quickly transferred into the simulated intestinal fluid, and the time-dependent change in the indomethacin concentration was measured for 12 h. The release rate was determined as the ratio of released indomethacin in the surrounding medium to the total indomethacin loaded in the capsules. The effects of the HPMCP content and drug loading amount in the capsules on the drug release behavior of the capsules were also studied. The concentrations of indomethacin in IF2 were set as 22.5, 45.0, and 65.0 mg·mL–1 . The concentrations of HPMCP in OF2 were set as 0, 0.0625, 0.1250, 0.2500, and 0.5000 mgmL–1 . Each experiment was repeated three times to obtain the mean values
《2.8. Cytotoxicity assay of the θ-ACPSi capsules》
2.8. Cytotoxicity assay of the θ-ACPSi capsules
A cell cytotoxic experiment was performed, mainly to detect the biocompatibility of the composite capsule wall materials. 3T3 cells (from the Stem Cell Bank, Chinese Academy of Sciences, China) and L929 cells (from the Kunming Cell Bank, Chinese Academy of Sciences, China) were cultured in DMEM containing 10% FBS, 100 units per milliliter penicillin, and 100 units per milliliter streptomycin in a constant temperature incubator at 37 °C and 5% CO2. The cytotoxicity of the θ-ACPSi capsules was evaluated by means of a CCK-8 assay. The capsule samples were ground with an agate mortar and then formulated into the suspensions used for the cytotoxicity experiment. 3T3 cells and L929 cells were separately seeded in 96-well plates; then, the θ-ACPSi capsule suspensions were added and diluted to different concentrations ranging from 50 to 2000 μg·mL–1 , and the cells were incubated for 12, 24, and 48 h, respectively. At predetermined time intervals, the medium of the 96-well plate was replaced with fresh medium plus 10 μL of CCK-8 solution. After 2 h of post-treatment incubation at 37 °C, the absorbance of each well was measured at 450 nm with a microplate reader (D2004W, Shanghai Meiyingpu, China). The cell viability (%) was calculated by Eq. (3):

where As, Ac, and Ab are the absorbance values of the sample group (with the medium containing cells, CCK-8, and capsule suspension), the control group (with the medium containing only cells and CCK8), and the blank group (with the medium containing only capsule suspension), respectively.
《2.9. In vivo drug release study of the θ-ACPSi capsules》
2.9. In vivo drug release study of the θ-ACPSi capsules
The in vivo release of indomethacin from the θ-ACPSi capsules was assessed using healthy New Zealand white rabbits (Chengdu Dossy Experimental Animals Co., Ltd., China) weighing (2.8 ± 0.2) kg. All procedures involving the animals were consistent with the guidelines of the Provisions and General Recommendation of Chinese Experimental Animals Administration Legislation, and were approved and supervised by the Ethics Committee of Chengdu Dossy Experimental Animals Co., Ltd. (approval number: DOSSY20190624002). Six rabbits were randomly divided into two groups, A and B (with three rabbits in each group). After 12 h of fasting with free access to water, each rabbit in group A was orally dosed with eight θ-ACPSi capsules at an indomethacin dose of 2.5 mg·kg–1 , while each rabbit in group B was orally perfused with a 2.5 mL CMC-Na suspension containing the same dosage of indomethacin. Blood samples were collected from the ear vein at predetermined time points (0, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0, 6.0, 8.0, and 12.0 h) and stored in heparinized tubes. Plasma samples were obtained via the centrifugation of blood samples at 4 °C for 10 min (4000 r·min–1 ), and the supernatants were frozen immediately and stored in heparinized tubes at –20 °C until the assay. For deamination of the indomethacin concentration, each plasma sample (0.1 mL) was mixed with methanol (0.3 mL) and, after centrifugation at 10 000 r·min–1 for 10 min, the supernatant was analyzed using high-performance liquid chromatography (HPLC; U3000, Thermo Scientific, USA) with a C18 column. A mixture of 0.24% acetic acid solution and acetonitrile (80/20, v/v) was used as the mobile phase at a flow rate of 1.0 mL·min–1 .
《3. Results and discussion》
3. Results and discussion
《3.1. Design strategy for the θ-shaped capsules》
3.1. Design strategy for the θ-shaped capsules
The fabrication procedures and the intestinal-targeted controlled-release mechanism of the proposed θ-shaped Caalginate-based capsules are schematically illustrated in Figs. 1 and 2. First, the θ-AC capsule is prepared by combining a coextrusion minifluidic technique [35] (Fig. 1(a)) with a complex coacervation method (Fig. 1(b)). The drug chamber of the θ-AC capsule is loaded with a hydrophobic drug, and the shell of the drug chamber is embedded with enteric HPMCP microspheres (Fig. 1(c)). The booster chamber encapsulates PAA polymers as the booster [38,39]. Second, protamine molecules are adsorbed onto the surfaces of the θ-AC capsules to fabricate the θ-shaped Ca-alginate–chitosan/protamine (θ-ACP) capsules (Figs. 1(d) and (e)), which are then coated with rigid silica layers using the biosilicification method to prepare the θ-ACPSi capsules (Figs. 1(f) and (g)) [15]. The Ca-alginate capsules have poor mechanical stability, so it is easy to swell and dissolve in the pH of the intestinal environment. In a weakly acidic environment (pH < 6), the chitosan is positively charged and the alginate is negatively charged. An alginate–chitosan complex capsule wall is formed by the complex coacervation of these two polyelectrolytes with opposite charges, which can improve the mechanical stability of the Ca-alginate capsule [9,30,34,40]. In this work, the amount of chitosan added is small, so there is still a large number of negative charges in the Ca-alginate network after the complex coacervation of 0.2 wt% chitosan and 2 wt% Na-alginate under Ca2+ crosslinking, and a large number of positively charged protamine molecules can still be adsorbed onto the capsule surfaces. Silicification can enhance the mechanical properties of the capsules to inhibit their swelling in the small intestine [16,41]. HPMCP microspheres with unique enteric dissolution characteristics rapidly dissolve in the digestive juices of the small intestine, but remain stable and insoluble in a solution with a pH of less than 5.5. The PAA (pKa 4.25) remains unchanged in the gastric juice (pH < pKa) but swells in the small intestine solution (pH > pKa), due to the electrostatic repulsion caused by deprotonization [38,39]. The boosting function of the booster agent is achieved by the osmotic and swelling pressures produced by the stretching of the PAA polymer chains at pH 6.8. Furthermore, the θ-ACPSi capsules exhibit pH-responsive permeability driven by the electrostatic interactions between the Caalginate networks and the protamine molecules, such that the diffusional permeability across the capsule is smaller at a lower pH [15,16]. Therefore, under the synthetic effect of the above factors, drugs loaded in θ-ACPSi capsules are protected from leaking or releasing in the stomach environment (Fig. 2(b)). In contrast, in the small intestine (pH 6.8), the enteric HPMCP microspheres in the drug chamber shell dissolve rapidly, opening a large number of ‘‘microchannels.” Meanwhile, the PAA in the boosting chamber swells, providing a driving force that pushes the hydrophobic drugs out through the ‘‘microchannels” at a constant release rate (Fig. 2(c)).
《Fig. 2》
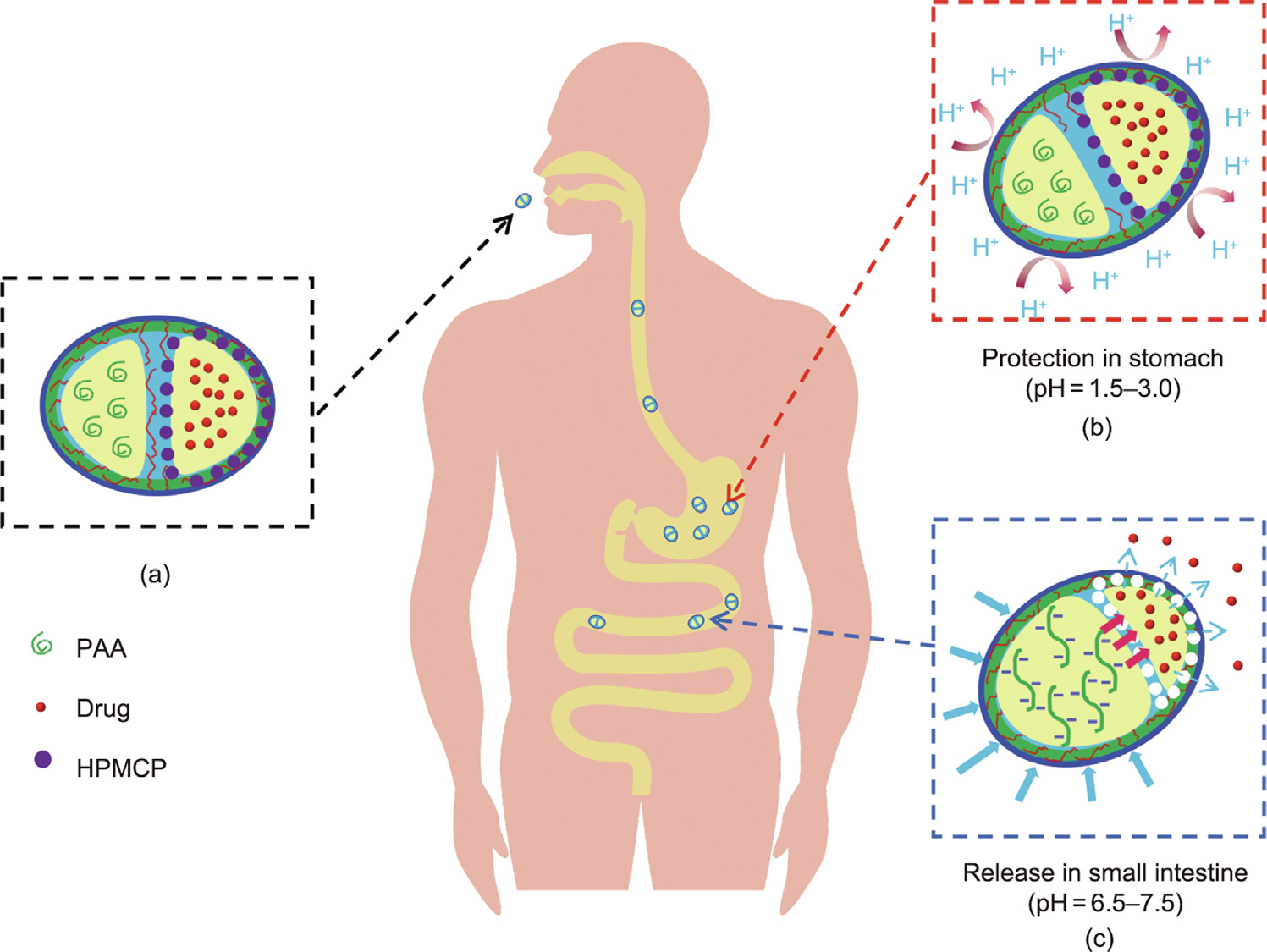
Fig. 2. Schematic illustration of the transportation of the intestinal-targeted θ-ACPSi capsules through the gastrointestinal tract. (a) θ-ACPSi capsules are loaded with indomethacin; (b) the θ-ACPSi capsules provide protection for indomethacin in the stomach; (c) release indomethacin rapidly in the small intestine.
《3.2. Pumping effect of the booster chamber》
3.2. Pumping effect of the booster chamber
The pumping capacity of PAA as a booster was confirmed by comparing the pH-responsive swelling behavior of singlechambered Ca-alginate–chitosan (o-AC) and Ca-alginate– chitosan/protamine/silica (o-ACPSi) capsules loaded with and without PAA under different pH conditions (Fig. 3). In this work, a phosphate buffer solution at pH 2.5 was used as simulated gastric fluid, and a phosphate buffer solution at pH 6.8 was used as the simulated intestinal fluid [6,10]. The PAA-loaded o-AC (PAA@oAC) capsules and o-ACPSi (PAA@o-ACPSi) capsules all showed good sphericity and uniform particle size (Figs. 3(a) and (b)). Before and after biosilicification, the capsules were transparent and white, respectively. Figs. 3(c) and (d) show the volume changes in the four kinds of capsules in different buffer solutions, and the dynamic swelling processes of these capsules when transferred from simulated gastric fluid to simulated intestinal fluid are shown in Fig. 3(e). All capsules were quite stable and did not swell at pH 2.5. Both of the o-AC capsules (i.e., with and without PAA) gradually swelled at pH 6.8, and the PAA@o-AC capsule became more transparent after 8 h (Fig. 3(c-iv)). The swelling ratio of the o-AC capsules with PAA was significantly higher than that without PAA. This difference in the swelling ratios of the capsules is simply due to the swelling effect of PAA, which demonstrates the driving force of PAA within the (simulated) small intestine at pH 6.8. In contrast, the o-ACPSi capsules hardly swelled at pH 6.8 due to the effective inhibition of the rigid silica layer [42].
《Fig. 3》
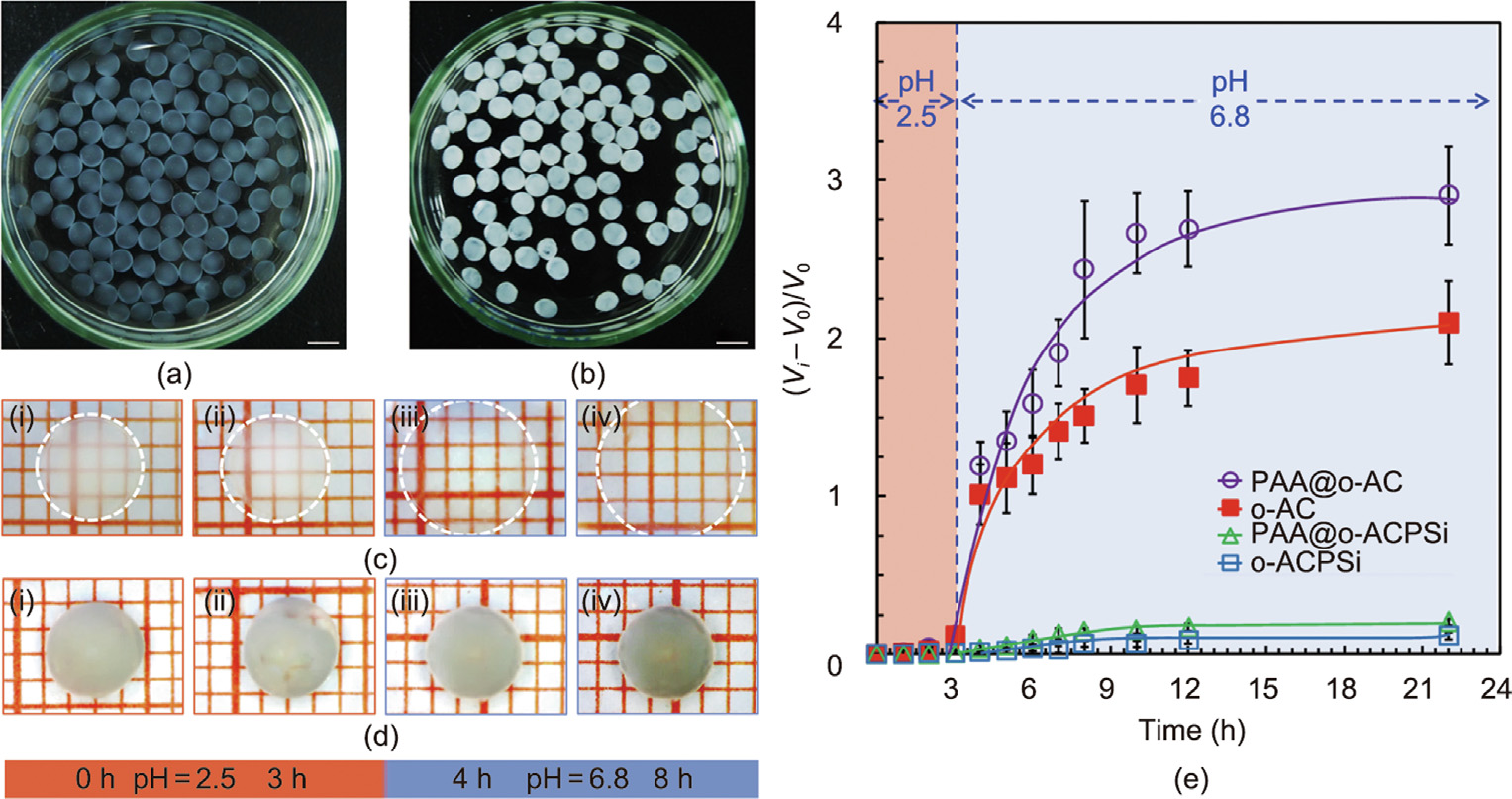
Fig. 3. (a, b) Optical images of (a) PAA-loaded o-AC (PAA@o-AC) capsules and (b) PAA-loaded o-ACPSi (PAA@o-ACPSi) capsules (scale bar is 5 mm). (c, d) Microscopic photographs of (c) one PAA@o-AC capsule and (d) one PAA@o-ACPSi capsule in solutions with different pH values and at different times. (e) Volume change rates of o-AC and o-ACPSi capsules loaded with and without PAA in solutions with different pH values.
《3.3. pH-responsive characteristics of HPMCP microspheres》
3.3. pH-responsive characteristics of HPMCP microspheres
Fig. 4(a) shows the formation process of the HPMCP microspheres with oil-in-water (O/W) emulsions, fabricated using microfluidic method. As the mixed solvent of dichloromethane and ethanol gradually diffuses into the external water phase, the droplet size gradually decreases and becomes stable after 15 min. Finally, HPMCP microspheres with an average particle size of about 52 μm are obtained (Fig. 4(b)). As shown in Figs. 4(c) and (d), the changes in the morphology and size of the HPMCP microspheres in pH 2.5 and pH 6.8 buffer solutions can be observed under a microscope. The prepared HPMCP microspheres have good sphericity and monodispersity, and the particle size remains almost unchanged in the pH 2.5 buffer solution. However, the HPMCP microspheres dissolve rapidly within 5 min in the pH 6.8 buffer solution, due to their excellent enteric solubility.
《Fig. 4》
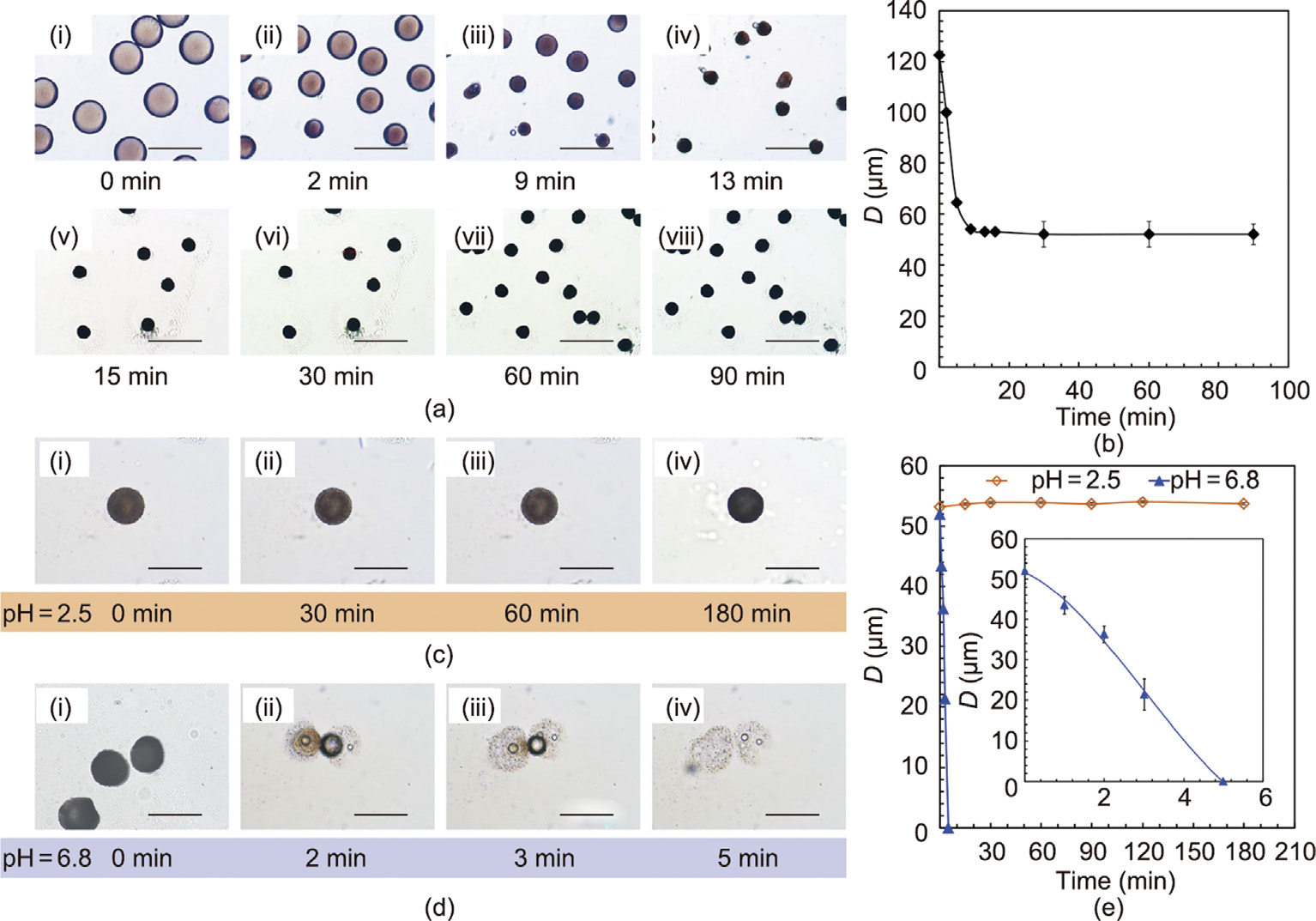
Fig. 4. pH-responsive performance of the HPMCP microspheres at pH 2.5 and pH 6.8. (a) Microscopic pictures and (b) particle size (D) change of HPMCP microspheres at different times during the formation process (scale bar is 200 μm). (c, d) Microscopic pictures and (e) particle size changes of the prepared HPMCP microspheres at different times at pH 2.5 and pH 6.8 (scale bar is 80 μm).
《3.4. Morphologies and microstructures of the θ-shaped capsules》
3.4. Morphologies and microstructures of the θ-shaped capsules
Optical images and CLSM images of the prepared θ-AC capsules and θ-ACPSi capsules without HPMCP and PAA in 0.2 moL·L–1 acetic acid solution are shown in Fig. 5. The θ-AC capsules and θ-ACPSi capsules all show a highly uniform spheroidal shape and a clear θ-shaped double-chambered structure (Figs. 5(a) and (e)). Rh-PAA (40 kDa) and LR300 with red fluorescence were respectively loaded into the booster chambers of the θ-AC capsules (Figs. 5(b)–(d)) and θ-ACPSi capsules (Figs. 5(f)–(h)) to further confirm the double-chambered structures of the capsules. When the molecular size of the solute is larger than the effective pore size of the Ca-alginate networks, the solute cannot penetrate the Ca-alginate capsule shell [17,43]. As shown in Figs. 5(d) and (h), the Rh-PAA and LR300 cannot diffuse into the drug chamber or booster chamber, confirming the existence of the separation membrane between the two chambers. The non-diffusion of Rh-PAA indicates that PAA with a molecular weight of 40 kDa can be effectively encapsulated within the booster chamber to avoid leakage and cross-contamination. Moreover, the central bulge of the separation membrane may be caused by shrinkage of the alginate chain and diffusion of Ca2+ [35]. In addition, after siliconization, the surface of the capsule becomes white and the internal structure cannot be observed.
《Fig. 5》
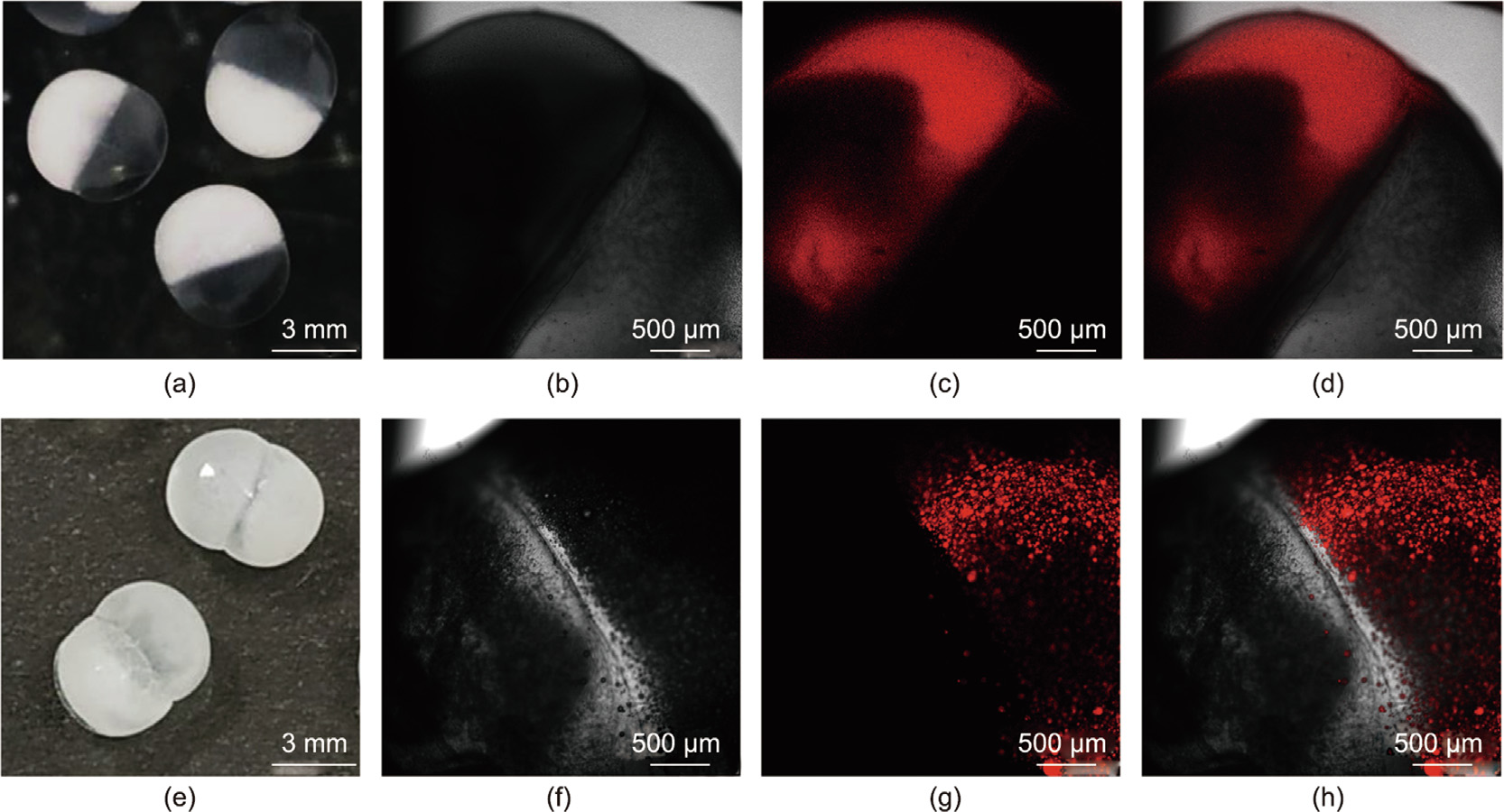
Fig. 5. θ-AC and θ-ACPSi capsules with two chambers for co-encapsulation. (a) Digital photograph and (b–d) CLSM photographs of θ-AC capsules with the booster chamber encapsulating Rh-PAA (red fluorescence). (e) Digital photograph and (f–h) CLSM photographs of θ-ACPSi capsules with the drug chamber encapsulating LR300 (red fluorescence). (b) and (f) are on transmission channels, (c) and (g) are on fluorescent channels, and (d) and (h) are on overlap channels.
SEM images of freeze-dried θ-ACPSi capsules with PAA and embedded with HPMCP microspheres are shown in Fig. 6. Before freeze-drying, the capsule sample was immersed into 0.2 moL·L–1 acetic acid solution. The θ-ACPSi capsule shows a good ellipsoidal shape (Fig. 6(a)), and the surface of the capsule shell is dense and smooth. Two separate hollow chambers can be clearly seen in the θ-ACPSi capsule (Fig. 6(b)), and the sections of the drug chamber shell and booster chamber shell both present a loose network microstructure coated with a dense silica outer layer (Fig. 6(b)). Embedded enteric HPMCP microspheres can be clearly seen in the drug chamber shell; the silica shell thickness is (21.2 ± 4.0) μm (Fig. 6(c)). However, there is no silica outer layer on the separation membrane between the booster chamber and the drug chamber (Fig. 6(d)). Because the protamine only adsorbs onto the outer surface of the θ-AC capsules, the subsequent silicification reaction only occurs on the outer surface of the θ-ACP capsules. That is, the outer capsule shell coated with a silica layer is rigid and can resist the swelling of the capsules to avoid deformation, while the separation membrane is elastic, so that it can be deformed by the swelling of PAA to squeeze the drug chamber and promote drug release.
《Fig. 6》

Fig. 6. SEM images of θ-ACPSi capsules. (a) An entire θ-ACPSi capsule; (b) crosssectional view of a θ-ACPSi capsule; (c) cross-sectional view of the drug chamber shell embedded with HPMCP microspheres; (d) cross-sectional view of the separation membrane between the drug chamber and the booster chamber.
SEM images of PAA-loaded θ-ACPSi capsules in a buffer solution of pH 6.8 after 8 h are provided in Fig. 7. The silica outer layer of the capsule still maintains a dense structure (Figs. 7(a) and (b)). In contrast, compared with Fig. 6(c), the separation membrane is deformed and bends toward the drug chamber due to the swelling of PAA (Fig. 7(c)), which confirms that the designed osmotic pumping occurred in a buffer solution of pH 6.8.
《Fig. 7》

Fig. 7. SEM images of θ-ACPSi capsules at pH 6.8 after 8 h. (a) An entire θ-ACPSi capsule; (b) surface; (c) cross-sectional view of a θ-ACPSi capsule; (d) crosssectional view of a θ-ACPSi capsule shell.
At pH 2.5, the diameter and shape of the θ-ACPSi capsules hardly changed due to the dense and stable network structure of the Ca-alginate [10,44] (Fig. S2 in Appendix A). However, pure Ca-alginate capsules typically swell and decompose in a high pH solution. When transferred into a medium at pH 6.8, the swelling rate of the θ-ACPSi capsules was controlled within 10% (Fig. S2). The rigid silica shell protects the stability of the internal Caalginate/protamine composite layer and inhibits swelling of the capsules in an intestinal environment, and the addition of chitosan improves the stability of the capsule to ensure the integrity of the capsules at higher pH, indicating the excellent stability of the θ-ACPSi capsules at high pH [9,30,34,40].
The stress–strain relationships of the capsules before and after silicification were studied by means of a pressure test. The capsule deformed under the compressive force (Figs. S3(a) and (b) in Appendix A). When the compressive force reached a certain value, the capsule was eventually damaged due to the rigid silica outer layer of the θ-ACPSi capsules and the fact that its elasticity is lower than that of the θ-AC capsules. Therefore, compared with the degree of deformation of the θ-AC capsules, the deformation degree of the θ-ACPSi capsules was reduced under compression, and the stress and strain values also decreased (Figs. S3(c)–(e) in Appendix A).
In practical applications, the prepared capsules can be stored after freeze-drying. If they are to be used within a short time after preparation, the capsules can be stored in a 0.2 moL·L–1 acetic acid solution. Due to the low permeability of the composite capsule wall in an acidic environment, it is difficult for the drug to diffuse from the capsules. The drug loading content of capsules stored in 0.2 moL·L–1 acetic acid solution for four weeks (28 d) remained almost unchanged, as shown in Fig. S4 in Appendix A. This result means that there is no drug leakage when the capsules are stored in an acetic acid solution.
《3.5. In vitro controlled drug release and cytotoxicity of the θ-ACPSi capsules》
3.5. In vitro controlled drug release and cytotoxicity of the θ-ACPSi capsules
Indomethacin, a typical non-steroidal anti-inflammatory drug, was used as the model hydrophobic drug in this work. The residence time of drug-loaded capsules in the gastrointestinal tract is 8–12 h [45], so the in vitro drug release time of the capsules was set at 12 h. To study the in vitro intestine-specific pumping release performance of the proposed θ-ACPSi capsules, the indomethacin-loaded capsules were first immersed into the simulated gastric fluid for 3 h and then transferred into the simulated intestinal fluid for 9 h, in order to simulate the transport process of the capsules through the human gastrointestinal tract. Fig. 8(a) shows the indomethacin release curves of θ-ACPSi capsules prepared with different HPMCP concentrations. These θ-ACPSi capsules are not loaded with PAA. In the simulated gastric fluid at pH 2.5, almost no indomethacin is released, due to the protective effect of the ACPSi capsule shell. At pH 2.5, the diffusion channels delineated by the electrically neutral Ca-alginate networks are ‘‘choked” by protamine molecules due to electrostatic repulsion between the positively charged protamine molecules [15,16]. In the simulated intestinal fluid at pH 6.8, due to the pH-responsive permeability of the hybrid capsule shell driven by the electrostatic interaction between the Ca-alginate networks and protamine molecules, indomethacin is rapidly released from the capsules. At pH 6.8, the Ca-alginate networks are negatively charged, and the positively charged protamine molecules are adsorbed onto the Ca-alginate networks due to electrostatic attraction, so the diffusion channels are in an ‘‘open” state [15,16]. Furthermore, due to the rapid dissolution of the HPMCP microspheres to open more ‘‘microchannels,” the indomethacin passes through the capsule shell more easily. With an increase in the HPMCP content in the capsule shell, the release rate of indomethacin increases.
Figs. 8(b)–(d) show the release curves for θ-ACPSi capsules with different drug loading concentrations. The drug release behaviors of the θ-ACPSi capsules with and without PAA or HPMCP are also compared. Similarly, almost no indomethacin is released from the four kinds of capsules (HPMCP@θ-ACPSi, PAA@θ-ACPSi, (HPMCP + PAA)@θ-ACPSi, and θ-ACPSi) in the simulated gastric fluid (pH 2.5), while it is gradually released in the simulated intestinal fluid (pH 6.8). Due to the dissolution of the enteric HPMCP microspheres at pH 6.8, the release rate of indomethacin from the HPMCP@θ-ACPSi capsules is greater than that from the θ-ACPSi capsules without HPMCP. Furthermore, due to the swelling of PAA at pH 6.8, which provides a pumping effect that promotes drug release, the release rate of indomethacin from the PAA@θ-ACPSi capsules is greater than that from the θ-ACPSi capsules without PAA. Under the synergistic effects of PAA and the HPMCP microspheres, the rapid release of indomethacin from the (HPMCP + PAA)@θ-ACPSi capsules is the fastest among the four kinds of capsules. The solubility of indomethacin in water is only 1.01 mg·mL–1 (37 °C, pH 6.8) [46]. Therefore, the indomethacin solutions in the drug chambers of the capsules are all suspensions. The cumulative drug release rate in this manuscript refers to the percentage of drugs released into the buffer solution out of the total amount of drugs encapsulated in the capsules. Since the drug solution in the capsules is a suspension, the release of indomethacin differs from a simple diffusion release driven by a drug concentration difference. Therefore, due to the calculation formula for the cumulative release rate, with an increase in the indomethacin loading content in the capsules, the cumulative release rate of indomethacin decreases. More importantly, the drug release from the (HPMCP + PAA)@h-ACPSi capsules reaches zero-order constant release. Therefore, the (HPMCP + PAA)@θ-ACPSi capsules present an excellent intestinal-targeting effect along with constant drug release characteristics.
《Fig. 8》
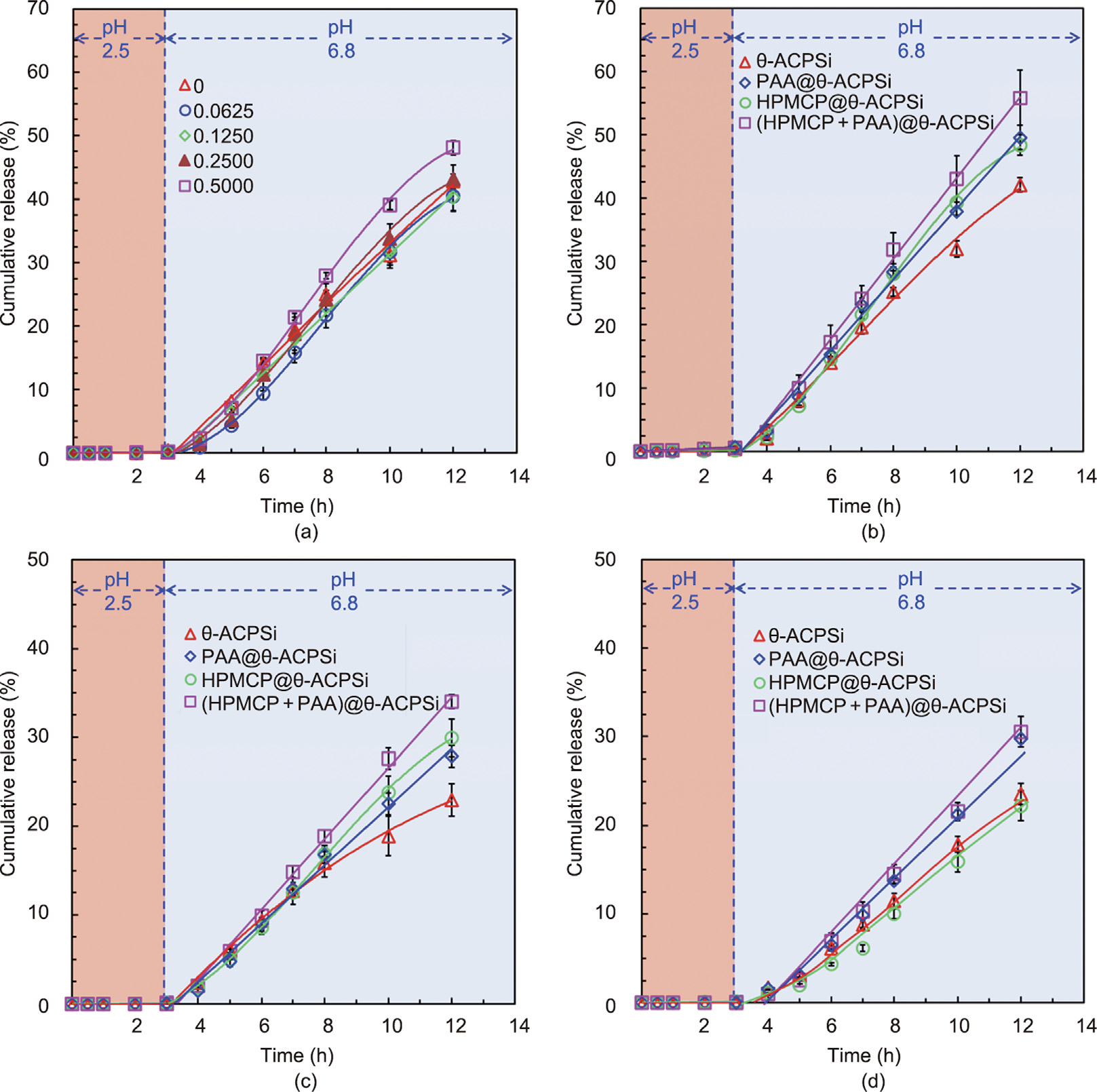
Fig. 8. Controlled-release behavior of indomethacin from different θ-shaped capsules in vitro. (a) Release curves for θ-ACPSi capsules with different contents (unit: mg·mL–1 ) of HPMCP microspheres in solutions with different pH values. (b)–(d) Release curves of different θ-shaped capsules (θ-ACPSi, PAA@θ-ACPSi, HPMCP@θ-ACPSi, and (HPMCP + PAA)@θ-ACPSi) with drug loading concentrations of (b) 22.5, (c) 45.0, and (d) 65.0 mg·mL–1 .
Fig. 9(a) shows the cumulative release rates of indomethacin from the four kinds of θ-ACPSi capsules at 12 h. With an increase in the loading concentration, the cumulative release rate of indomethacin decreases. The absolute release amounts of indomethacin from different capsules increase as the loading concentration increases, especially for the (HPMCP + PAA)@θ-ACPSi capsules (Fig. 9(b)). Because of the double effects of the ‘‘pumping force” and the ‘‘microchannel switch” at the different loaded indomethacin concentrations of (HPMCP + PAA)@θ-ACPSi capsules, the cumulative release rate and absolute release rate of indomethacin at 12 h are improved.
《Fig. 9》
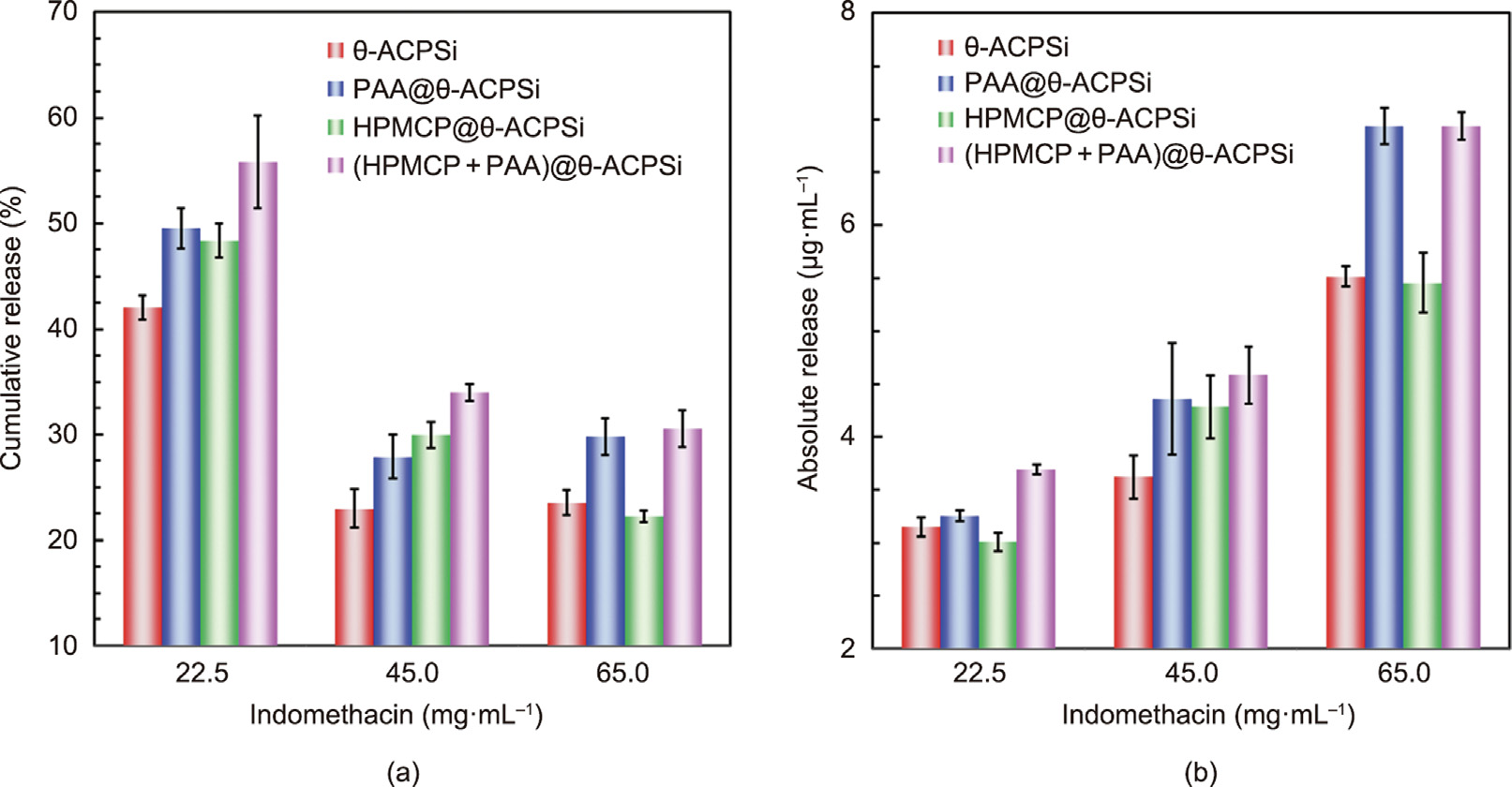
Fig. 9. (a) Cumulative release rates and (b) absolute release amounts of indomethacin from different θ-shaped capsules at 12 h.
《3.6. Cell cytotoxicity and the in vivo drug release of the θ-ACPSi capsules》
3.6. Cell cytotoxicity and the in vivo drug release of the θ-ACPSi capsules
Cell cytotoxicity assays of θ-ACPSi capsules with different concentrations ranging from 50 to 2000 μg·mL–1 were performed using 3T3 cells and L929 cells, and the results are shown in Fig. 10. For the 3T3 cells, there was nearly no loss of cell viability after 48 h of treatment with θ-ACPSi capsules with different concentrations (Fig. 10(a)). Even when the concentration of θ-ACPSi capsules reached 2000 μg·mL–1 , the cell viability of the 3T3 cells after 48 h was close to 100%. The cell viability of the L929 cells decreased slightly with an increase in the capsule concentration when the incubation time was increased from 12 to 48 h (Fig. 10(b)), but the loss of cell viability was still acceptable. These results indicate that the prepared θ-ACPSi capsules have good biocompatibility and hold potential for use as safe carriers for drug delivery.
《Fig. 10》
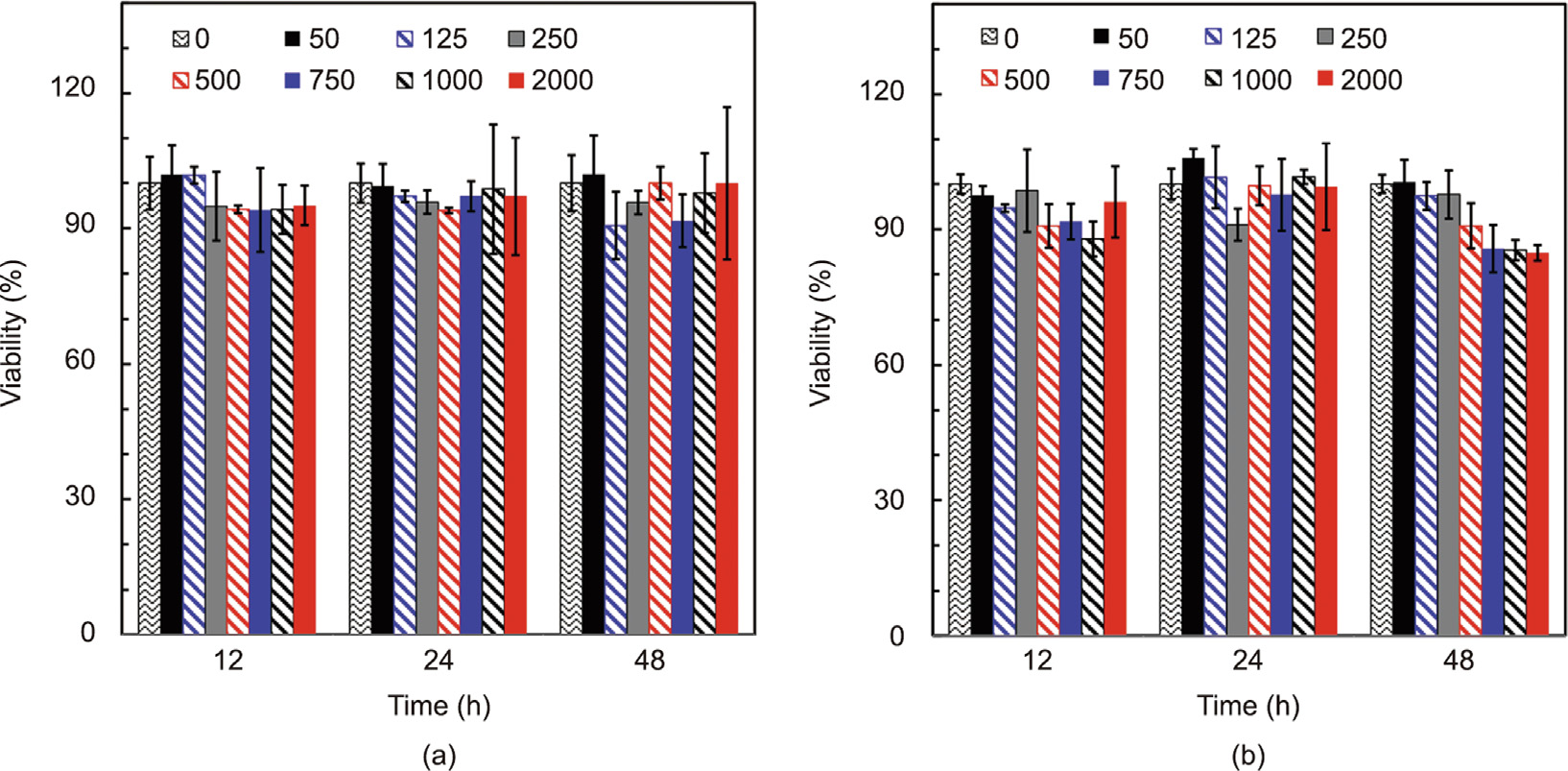
Fig. 10. Cell cytotoxicity of θ-ACPSi capsules. Viabilities of (a) 3T3 cells and (b) L929 cells treated with θ-ACPSi capsules at different concentrations for 12, 24, and 48 h. The concentration unit of the θ-ACPSi capsules is μg·mL–1 .
The effects of the intestinal-targeted delivery and the pumpingcontrolled constant release of the θ-ACPSi capsules were also evaluated by in vivo pharmacokinetic studies in New Zealand white rabbits, which compared the indomethacin release from θ-ACPSi capsules after oral administration with the direct oral perfusion of indomethacin suspension (Fig. 11). When treated with indomethacin suspension, the rabbit plasma concentration of indomethacin rapidly reached the maximum plasma concentration (Cmax) of 193 ng·mL–1 at 2 h (time to Cmax is defined as Tmax). However, when treated with the indomethacin-loaded θ-ACPSi capsules, the rabbit plasma concentration of indomethacin slowly increased at first, with a Cmax of 247 ng·mL–1 at 6 h. That is, the Tmax of the indomethacin from the θ-ACPSi capsules was delayed by 4 h, indicating that the θ-ACPSi capsules have a good intestinal-targeting effect. Furthermore, the plasma concentration of indomethacin for rabbits treated with the indomethacinloaded θ-ACPSi capsules was maintained at a higher level for a long time (Fig. 11(b)). The area under curve (AUC) value of the plasma concentration of indomethacin for rabbits treated with the indomethacin-loaded θ-ACPSi capsules was 1.63 times higher than that for rabbits treated with indomethacin suspension, which means that the intestinal-targeted delivery of the θ-ACPSi capsules provided a better indomethacin-absorption effect.
《Fig. 11》
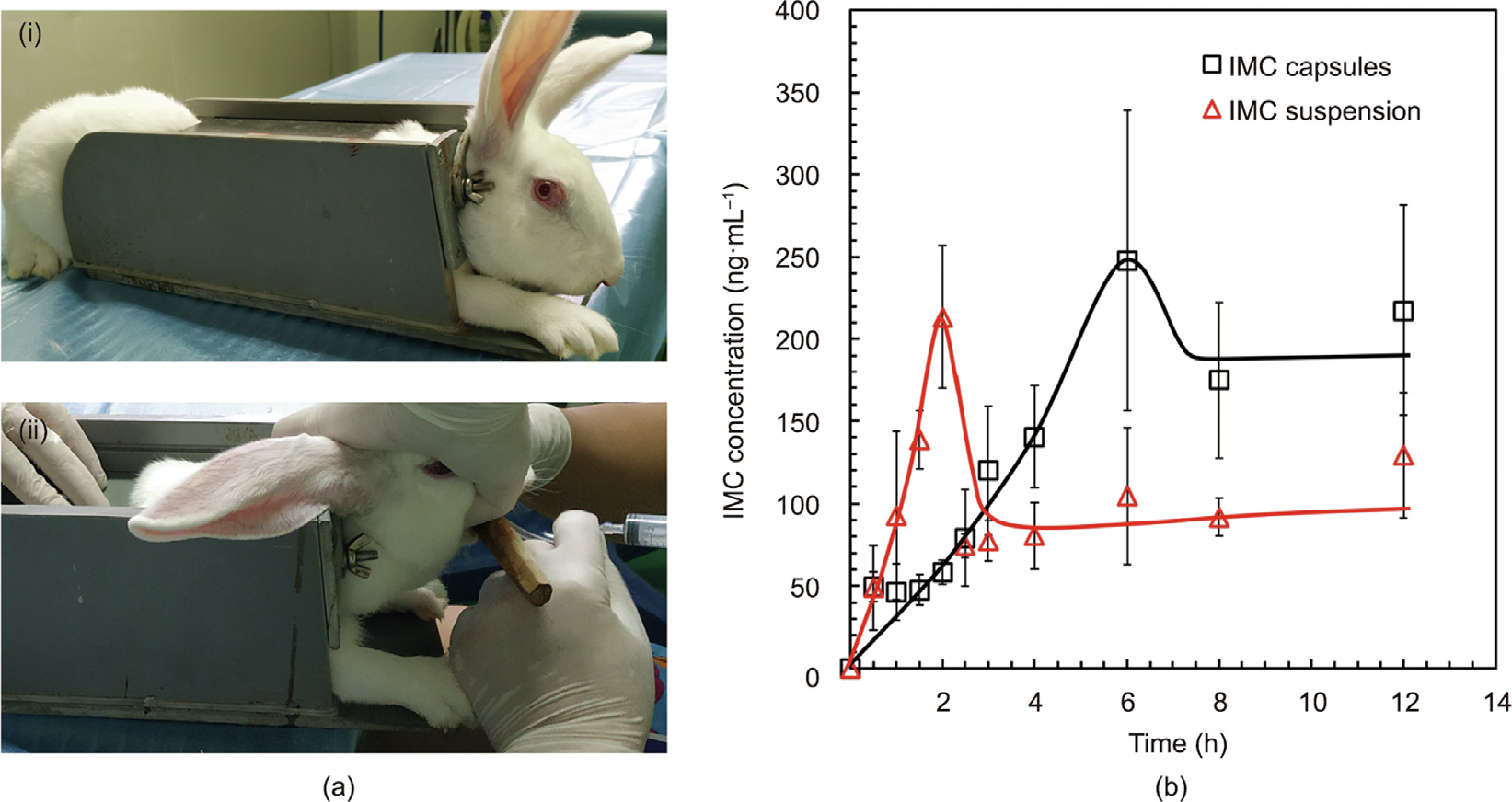
Fig. 11. In vivo drug release from θ-ACPSi capsules with a New Zealand white rabbit as the animal model. (a-i) Immobilization and (a-ii) administration to rabbit; (b) plasma concentration–time profiles of indomethacin (data represent mean ± standard deviation, n = 3). IMC: indomethacin.
《4. Conclusions》
4. Conclusions
A novel intestinal-targeted θ-shaped Ca-alginate-based capsule with pumping effects was successfully developed for the controlled release of hydrophobic drugs in the treatment of intestinal diseases. The proposed θ-ACPSi capsule not only offers improved protection for drugs in the stomach environment but also has excellent intestinal-targeted characteristics to ensure the release of indomethacin in the small intestine. When the proposed indomethacin-loaded θ-ACPSi capsules enter the stomach, which has an extremely low pH, the diffusion channels in the composite shells are choked with protamine molecules, so that the encapsulated indomethacin is not released. After the proposed capsules enter the small intestine, which has a pH of 6.8, the HPMCP microspheres in the shell of the drug chamber dissolve, opening ‘‘microchannels” for the release of the encapsulated indomethacin, and the PAA contained in the booster chamber swells to provide impetus to push out the encapsulated indomethacin. With the synergistic action of these two effects, the intestinal-targeted controlled release of hydrophobic drugs is achieved. Compared with treatment with indomethacin suspension, the time of peak concentration of indomethacin in the plasma of rabbits treated with indomethacin-loaded θ-ACPSi capsules was delayed by at least 3 h, and the AUC value of the plasma concentration of indomethacin for rabbits treated with the indomethacin-loaded θ-ACPSi capsules was 1.63 times higher. The proposed intestinal-targeted θ-ACPSi capsule provides a novel potential model for responsive pumping controlled-release systems and intestinal-targeted drug delivery systems.
《Acknowledgments》
Acknowledgments
The authors gratefully acknowledge support from the National Natural Science Foundation of China (22078202 and 21991101).
《Authors’ contribution》
Authors’ contribution
Shuang Wen: conceptualization, investigation, methodology, validation, formal analysis, writing—original draft, review and editing, and visualization. Xiao-Jie Ju: conceptualization, resources, writing—review and editing, supervision, project administration, and funding acquisition. Wen-Ying Liu: formal analysis, data curation, and visualization. Yu-Qiong Liu: methodology and software. Xing-Qun Pu: conceptualization, investigation, methodology, validation, and formal analysis. Zhuang Liu: investigation, methodology, and resources. Wei Wang: methodology, conceptualization, and resources. Rui Xie: project administration and resources. Yousef Faraj: writing—review and editing. Liang-Yin Chu: conceptualization, resources, writing—review and editing, supervision, and funding acquisition.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Shuang Wen, Xiao-Jie Ju, Wen-Ying Liu, Yu-Qiong Liu, Xing-Qun Pu, Zhuang Liu, Wei Wang, Rui Xie, Yousef Faraj, and Liang-Yin Chu declare that they have no conflict of interest or financial conflicts to disclose.
《Appendix A. Supplementary data》
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2022.05.021.











 京公网安备 11010502051620号
京公网安备 11010502051620号




