

《1. Introduction》
1. Introduction
Polyester plastic is one of the most widely used types of plastic products in daily life [1–5]. In 2013 alone, approximately 5.6 × 107 t polyester plastic were produced, while less than 30% of the total amount was recycled [6]. The main plastic recycling methods include chemical and biological methods. However, chemical degradation of plastic requires high-temperature [7–9] and high-pressure [10–12] conditions and consumes a large amount of energy. In addition, this approach generates a large amount of substances that are toxic and harmful to the environment, and has the potential to cause secondary pollution [7,13]. By contrast, the biodegradation method appears to be milder and more environment friendly, and thus provides a new approach for the recovery of plastic products [7,14]. Previous research has mainly employed cutinases derived from Thermobifida fusca (T. fusca) [15], Fusarium solani pisi [16], and T. cellulosilytica [17]; these were able to degrade PET to some extent at 50 °C, but were inefficient at room temperature. Thus, efficient plastic degradation by an applicable normal-temperature bioprocess has remained a challenge. In 2016, Yoshida et al. [18] isolated a bacterium that produced two enzymes, poly(ethylene terephthalate) hydrolase (PETase) and mono-(2-hydroxyethyl) terephthalate hydrolase (MHETase), that were capable of degrading poly(ethylene terephthalate) (PET) to terephthalate (TPA) and ethylene glycol (EG) at 30 °C. Notably, PETase was shown to have a significantly higher ability to degrade PET at 30 °C than the previously reported enzymes LCC, TfH, and THc_Cut1 [18]. Thus, PETase was considered to be a potentially promising tool for plastic degradation.
Until now, the promising ability of the PETase enzyme has not been realized in applications. In order to improve the polyester plastic-degrading activity of PETase, a modification of the protein by rational design—which has been proven to be effective in enhancing the activities of previously used cutinases—was carried out. Herrero Acero et al. [19] almost tripled the activity of the cutinase from T. cellulosilytica by site-directed mutagenesis. In a similar study by Wei et al. [20], mutagenesis was used to increase the activity of the cutinase derived from T. fusca by 2.7 times while simultaneously mitigating product inhibition by mono-(2-hydroxyethyl) terephthalate. Silva et al. [21] modified the active site of Tfu_0883 to increase the hydrophobicity and/or create space; this strategy also successfully improved the enzymatic degradation of PET.
The rational design of proteins requires less screening than random mutagenesis and directed evolution. However, rational design often requires saturation mutation of the protein [22,23]. As PETase is located in the cell, it cannot come into contact with the solid PET, which makes it impossible to establish a highthroughput screening method. Therefore, all of the mutants must be expressed in Escherichia coli (E. coli) and isolated, which is very time-consuming work [22]. Preliminary screening by a simple and convenient high-throughput technique has been a great challenge for researchers [23,24]. The emergence of cell-free proteinexpression systems provides an efficient and convenient method for enzyme studies. Compared with traditional intracellular expression in E. coli, cell-free protein-expression systems significantly shorten the expression time and reaction volume, thereby greatly supporting high-throughput screening [25]. Murthy et al. [25] used a 50 μL cell-free protein system to achieve the highthroughput synthesis of 63 proteins from Pseudomonas aeruginosa within 4 h, and 51 proteins were successfully expressed. Sawasaki et al. [26] developed a cell-free protein-expression system based on wheat seeds that can translate at least 50 genes in parallel. This system bypasses many of the time-consuming cloning steps of conventional expression systems, making it suitable for proteins. The high-throughput cell-free protein-expression system developed by Goshima et al. [27] was used to express 13 364 human proteins, 77% of which had biological activity. Therefore, the cell-free protein-expression system provides a platform for high-throughput protein expression, and cell-free protein synthesis breaks through the limitations of the cell wall. Consequently, the expressed protein can come into direct contact with solid PET, thereby providing the possibility of high-throughput screening of PET hydrolytic enzymes.
In this study, we analyzed the interaction between PETase and the substrate (a dimer of the PET monomer ethylene terephthalate, 2PET), by molecular docking. Modification of hot-spot amino acids around the substrate-binding groove was carried out by genetic engineering. At the same time, high-throughput expression and preliminary screening of these proteins were performed using the cell-free protein-expression system, and mutations at three points were found to result in an increase in PETase activity. The resulting mutants exhibited greater catalytic efficiency in the degradation of a PET film than wild-type PETase. During the verification of in vitro enzyme activity parameters, the mutants demonstrated strong catalytic activity and a high substrate-conversion rate. In addition, the ability of high-throughput screening methods to quickly provide mutants with enhanced enzymatic activity through the rational design of PETase was verified.
《2. Materials and methods》
2. Materials and methods
《2.1. Mutation design》
2.1. Mutation design
A three-dimensional model of PETase, the active site, and the key amino acids related to catalytic activity have been established in previous studies [28]. The active site of PETase was mutated according to the model in order to reduce the steric hindrance around the active site and enhance the affinity between the active site and polyester plastic, thus allowing PETase to bind and react with PET more efficiently.
The active site of cutinases and lipases is a highly conserved sequence, -G-X1-S-X2-G-, which has been found in LCC [29], TfCut2 [30], and TfH [31]. By comparing the sequence of PETase with the sequence of TfCut2, we found that the catalytic triad of PETase consisted of S131, H208, and D177. Mutation was performed to create space around the active site, to increase the hydrophobicity of the amino acids around the active site, or to improve the affinity of the amino acids around the active site for PET. The principle of substitution was used in replacing the original amino acids with smaller or more hydrophobic amino acids.
《2.2. Site-directed mutagenesis of PETase》
2.2. Site-directed mutagenesis of PETase
The PETase gene was synthesized at GENEWIZ® (Suzhou, China) after codon optimization of the sequences for their modified expression in E. coli. The flexiblepeptide (GGGGS)3 was introducedat the Cterminus of PETase during the synthesis process. The BamHI/NcoI restriction enzyme site was added to both ends of the PETase. PETase was ligated into the pRSET-CFP vector using BamHI/NcoI and fused to cyan fluorescent protein (CFP). pRSET-PETase-CFP was constructed as a template for the site-directed mutagenesis of PETase. Site-directed mutagenesis of PETase was performed using the fast site-directed mutagenesis kit (KM101, TIANGEN Biotech Co., Ltd., China), and was then verified by sequencing. The megaprimers used to site-mutate PETase are listed in Table S1.
《2.3. Protein expression in the cell-free protein synthesis system》
2.3. Protein expression in the cell-free protein synthesis system
Recombinant PETase mutants were expressed in the E. coli-based cell-free protein synthesis system [32]. The protocol of crude extract preparation and batch mode cell-free reaction is described in Ref. [33]. pRSET-CFP (50 μL per reaction system) was used as the carrier for cell-free protein synthesis at 37 °C for 12 h; the reaction was monitored in real time using a microplate reader. The fluorescence detector was set at an excitation wavelength of 435 nm and an emission wavelength of 479 nm.
《2.4. Preliminary screening of mutations》
2.4. Preliminary screening of mutations
Solution buffer (50 mmol·L-1 bicine-NaOH, pH 8.5) was added to each cell-free protein-expression system in order to dilute the fluorescence intensity to 1000; 10 lL of the resulting solution was then pipetted into the 1990 μL (50 mmol·L-1 bicine-NaOH, pH 8.5) reaction system. Next, a 1.5 cm × 1.0 cm PET film (Goodfellow, 577-529-50) was added and reacted at 30 °C for 48 h. The reaction was terminated by dilution of the aqueous solution with 1 mol·L-1 NaOH followed by heat treatment (50 °C, 10 min). After centrifuging at 12 000 r·min-1 for 1 min, 200 μL of supernatant was pipetted into a 96 well plate, and the absorbance at 240 nm was measured with a microplate reader [15]. Before addition, the PET film was prewashed with 1% sodium dodecyl sulfate (SDS), ethanol, and distilled water at 50 °C for 30 min, and then dried for 48 h at 50 °C. All experiments were carried out in triplicate. Controls were performed with CFP instead of with the PETaseCFP fusion protein.
《2.5. Protein expression and purification in E. coli》
2.5. Protein expression and purification in E. coli
All of the wild-type and mutant proteins were cloned into the pET28a vector and transformed into E. coli BL21 (DE3) cells that were grown in Luria-Bertani (LB) medium at 37 °C to an OD600 from 0.6 to 0.8. Using the plasmid that was constructed in the cell-free protein synthesis as a template, the wild-type PETase and R61A, L88F, and I179F were cloned by polymerase chain reaction (PCR). BamHI and EcoRI sites were introduced at both ends of the cloned gene and were cloned into pET28a by restriction endonuclease digestion. Afterward, the culture was cooled to 16 °C and induced with isopropyl β-D-thiogalactopyranoside (IPTG) at a final concentration of 0.1 mmol·L-1 for 20 h at 16 °C and 160 r·min-1. The cells were harvested by centrifugation (25 min, 10°C, 4000 r·min-1). Cell pellets from the 200 mL cell culture were re-suspended in 10 mL of Ni-NTA lysis buffer (50 mmol·L-1 NaH2PO4, 300 mmol·L-1 NaCl, 10 mmol·L-1 imidazole, pH 8). The cells were disrupted by sonication on ice, and the lysates were centrifuged (30 min, 10°C, 4000 r·min-1). The supernatant was applied to a His-Tagged nickel column (Beyotime® Biotechnology, China). After washing the unbound protein with 10 mL of lysis buffer, the bound protein was eluted with elution buffer (50 mmol·L-1 Tris-HCl, pH 7.5, 300 mmol·L-1 NaCl, 200 mmol·L-1 imidazole). Buffer exchange to 50 mmol·L-1 Na2HPO4-HCl (pH 7.0) and 100 mmol·L-1 NaCl solution was then carried out using a PD-10 gel filtration column (GE Healthcare, USA).
《2.6. Kinetic studies》
2.6. Kinetic studies
Activity measurements were conducted using para-nitrophenyl acetate (PNPA) as a substrate to analyze the Michaelis kinetic parameters, as previously described [29,33,34]. The kinetic parameters were determined at 30°C and pH 7.0 with substrate concentrations ranging from 0.5 to 8.0 mmol·L-1. All experiments were carried out in triplicate. Kinetic data were calculated with Sigma Plot 11.0 Software (Systat Software GmbH, Erkrath, Germany) utilizing the Double-Reciprocal Plot. The kinetic parameters and the calculation process are shown in the Appendix A (Fig. S1 and Table S2).
《2.7. Enzymatic hydrolysis of PET》
2.7. Enzymatic hydrolysis of PET
Prior to PET hydrolysis, all of the films were washed in three consecutive steps, each of which lasted 30 min: First, they were washed in a solution of 1% SDS, next with ethanol, and finally with deionized water. A PET film (1.5 cm × 1.0 cm) was placed in a reaction solution containing 5 μg of PETase and 1.5 mL of 50 mmol·L-1 bicine-NaOH (pH 8.5), and reacted for 48 h at 30°C. The reaction was terminated by dilution of the aqueous solution with 1.0 mol·L-1 NaOH followed by heat treatment (50°C, 10 min). The prewashed PET films were washed, dried, and subjected to gravimetric weight-loss determination. All experiments were carried out in triplicate. Controls were performed using 1.5 mL of bicine-NaOH (pH 8.5) without enzyme.
《2.8. Scanning electron microscopy》
2.8. Scanning electron microscopy
After the reaction, the experimental group and the control group were washed and dried, and then observed with a scanning electron microscope. The relevant places in the samples were scanned using an electron microscope at 2500× magnification.
《3. Results》
3. Results
《3.1. Structural modeling of PETase for the selection of mutation sites》
3.1. Structural modeling of PETase for the selection of mutation sites
The active site of PETase and the amino acids associated with its catalytic activity were identified in a previous study [28]. The three-dimensional structure of PETase approximately determined Y58 and M132 to be the oxyanion hole, and S131, D177, and H208 to be the catalytic triad. All the experiments were designed based on the three-dimensional model of PETase (Protein Data Bank code: 5XG0).
A crystal structure of PETase was created as a complex of the enzyme with the model substrate 2PET (Fig. 1). The oxyanion hole formed by Y58 and M132 is the catalytic core site for PETase. S131 participates in the cleavage of the ester bond as a nucleophile, and Y58, which contains aromatic groups, is necessary to bind the substrates. W156 and W130, which are located inside the substrate channel, are also involved in substrate binding. An analysis of the parts of the structure related to the binding between PETase and the substrate led to the choice of six amino acids around the active site for mutation: R61, L88, S178, I179, S209, and A211 (Fig. 1); all of these participate in substrate binding.
《Fig.1》

Fig. 1. Complex structural model of PETase and 2PET. The surface structure is colored in gray and the docked 2PET is colored in green. The substrate 2PET binding pocket is indicated with a black dotted line, and the key residues involved in the substrate-binding pocket are colored in yellow.
R61 is located at the end of the substrate; the hydrophobicity of the amino acid at this site affects the binding of the protein to the substrate. At the same time, the size of the modified amino acid affects the release of degradation products. Therefore, the arginine was mutated to an amino acid with high hydrophobicity and the lowest possible molecular weight. The amino acid alanine increases the contact between the enzyme and the substrate and facilitates the release of the substrate. L88 and S209 are located on the outside of the substrate-binding channel. Increasing the hydrophobicity of the changed amino acid can increase the affinity of the PETase to the substrate, making it easier for the protein to come into contact with PET. Therefore, S178 was mutated to phenylalanine, which has a stronger affinity with PET, and S209 was mutated to the more hydrophobic valine and the more hydrophilic phenylalanine, respectively, in order to enhance the activity of the enzyme. I179 is adjacent to the active site. Both steric hindrance and the hydrophobicity of this site can affect the catalytic activity of the enzyme; therefore, isoleucine was mutated into phenylalanine to increase the affinity of the enzyme to the substrate. In addition, isoleucine was mutated into valine; this does not reduce the hydrophobicity of the protein, but it does reduce the steric hindrance of the substrate binding site. A211 is located at the site of the substrate release. Too much hydrophobicity or too much steric hindrance of the amino acid at the site causes the substrate to be difficult to release. Therefore, arginine was mutated into a hydrophilic amino acid with the least steric hindrance—that is, proline—in order to promote the release of the substrate. S178 is located around the active site. Increasing the hydrophobicity of the amino acids can increase the affinity of the enzyme with the substrate; however, the S178 site is located in the center of the active site. In order to ensure that the activity of the enzyme was not affected, serine, a hydroxyl amino acid, was mutated into more hydrophobic hydroxyl amino acids.
《3.2. Screening of the PETase variant using cell-free protein synthesis》
3.2. Screening of the PETase variant using cell-free protein synthesis
PETase was expressed in the cell-free system (Fig. S2), and the fluorescence intensities of the CFP fused at the C-terminus were identified (Fig. 2(a)). The CFP fluorescence intensities of the proteins were distributed between 1400 and 12 000, and L88F, I179V, and I179F had the lowest values, which were less than 2000. The expression levels of S178T and S209V were relatively high, and their CFP fluorescence intensities reached 10 000 (Fig. 2(a)).
《Fig.2》
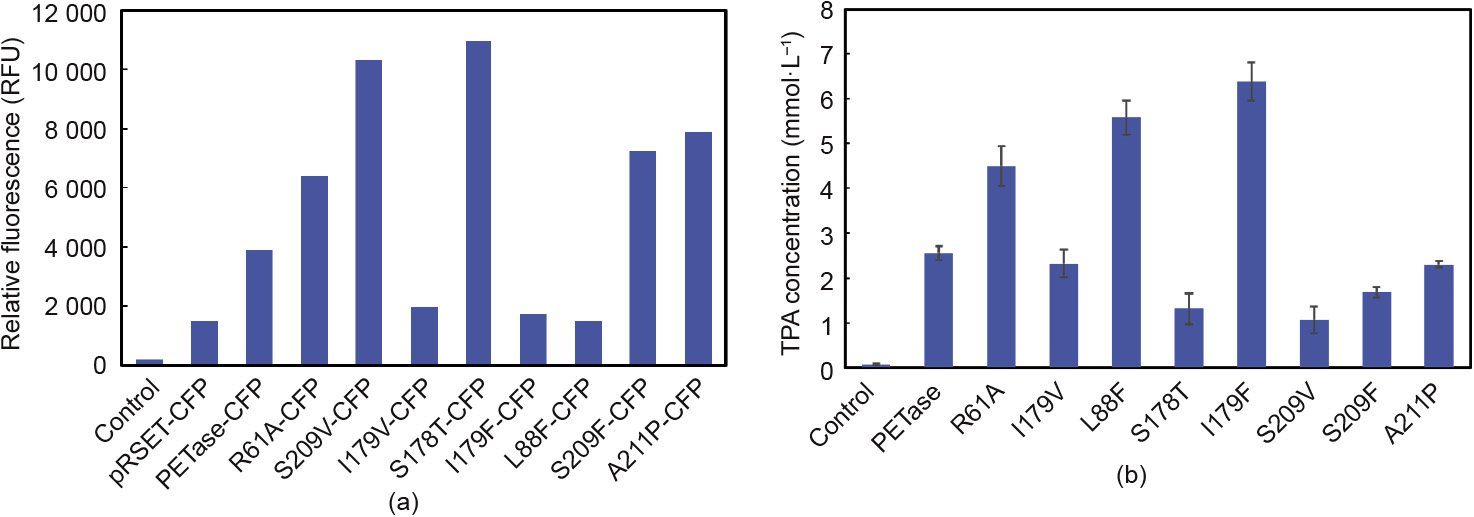
Fig. 2. Screening of a PETase variant using cell-free protein synthesis. (a) Relative fluorescence of CFP expressed in cell-free protein synthesis for 12 h; (b) at the same protein concentration, the TPA concentration of PETase and its variants after reacting with PET for 48 h at 30°C. RFU: relative fluorescence units.
A PET film was added to the reaction system and reacted at 30°C for 48 h. After dilution, the absorption peak at 240 nm was measured. The concentration of TPA was determined by establishing a standard curve between TPA concentration and OD240. Fig. 2(b) shows that among the mutants, the mutation of I179F has the strongest enzymatic activity—2.5 times greater than the activity of wild-type PETase—with 6.38 mmol·L-1 of TPA being released. Compared with the wild-type PETase, L88F hydrolyzes PET to TPA. The amount released was 5.38 mmol·L-1—2.1 times greater than the amount released by wild-type PETase. The enzymatic activity of R61A was 1.6 times greater than the activity of wild-type PETase. In contrast, the activity of the S178T and S209V mutants was reduced to only 29.7% and 38.2%, respectively, of the activity of wild-type PETase. The remaining mutants showed no significant increase or decrease in enzymatic activity.
《3.3. Verification of PET film degradation by the PETase variants》
3.3. Verification of PET film degradation by the PETase variants
To validate their ability to degrade PET, three variants (R61A, L88F, and I179F) were purified from E. coli, and the purified enzymes were reacted with PET films. The activity of the enzymes was characterized by quantifying the weight loss of the PET film. The degradation rates of the PET film following enzymatic degradation at 30°C for 48 h by PETase and its variants are shown in Fig. 3. Variant L179F shows the strongest catalytic activity; the weightloss rate of the PET film reached 22.5 mg per μmol·L-1 PETass per day. The catalytic activity of the L88F and R61A mutants was also improved compared with that of wild-type PETase: The degradation rate of wild-type PETase is 8.2 mg per μmol·L-1 PETase per day, whereas those of L88F and R61A were 17.5 and 13.5 per μmol·L-1 PETase per day, respectively.
《Fig.3》
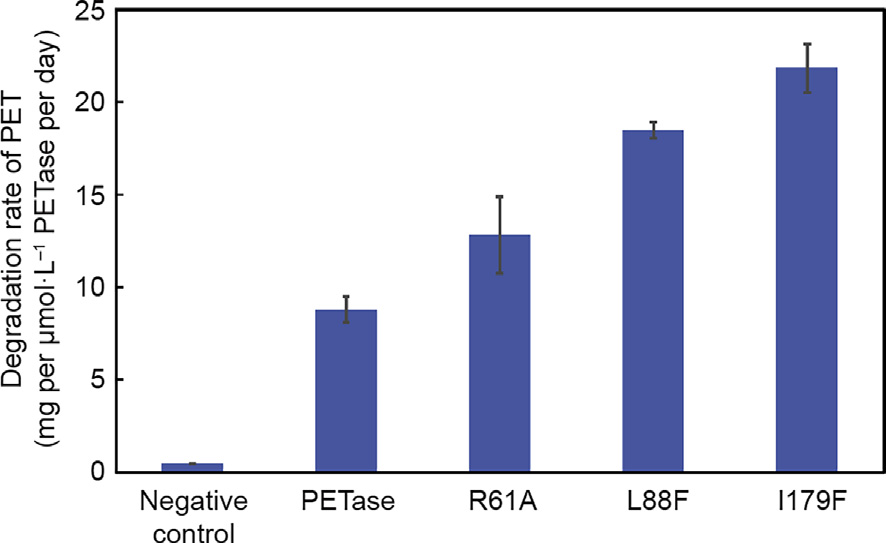
Fig. 3. Weight-loss rate of the PET film with PETase and its variants. Error bars indicate the standard deviation based on triplicate determinations.
In order to verify the changes in enzyme and substrate interactions, a complete kinetic study was undertaken, in which the performance of the mutants (R61A, L88F, and I179F) was compared with that of wild-type PETase. The soluble substrate PNPA was used to characterize and compare the differences in the Michaelis parameters between each variant and wild-type PETase (Fig. S1, Table 1). Among the three mutants that were initially screened for their increased enzymatic activity, the Michaelis constant, KM, values for L88F and R61A were (3.8 ± 0.4) and (4.5 ± 0.1) mmol·L-1, respectively; these values were similar to that of wild-type PETase ((4.6 ± 0.5) mmol·L-1). However, the mutation in I179F resulted in a decrease in the KM of PETase to just 1.2 mmol·L-1.
《Table 1》
Table 1 Kinetic parameters of PETase and its variants with PNPA.
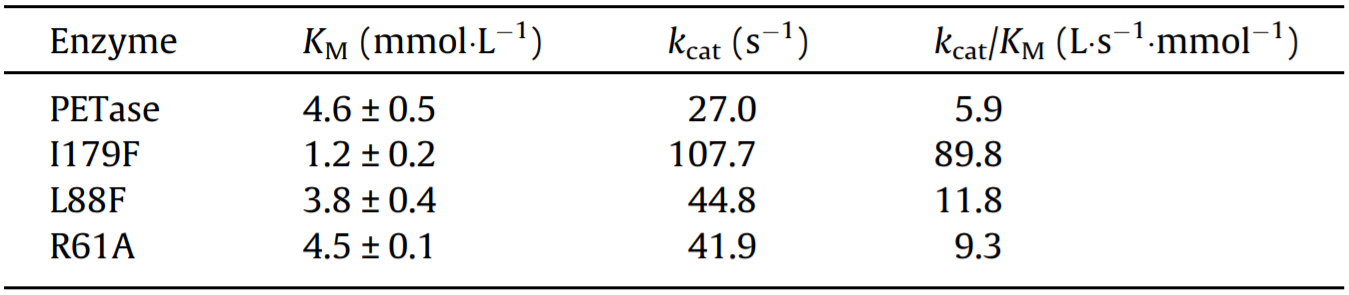
The constant kcat of wild-type PETase for catalyzing PNPA hydrolysis was calculated to be only 27.0 s-1. The kcat values of all the mutants selected from the primary screening were higher than that of wild-type PETase, with the kcat of I179F increasing by nearly four-fold, from 27.0 s-1 to 107.7 s-1. At the same time, the kcat/KM of I179F increased to 89.8 L·s-1·mmoL-1, which is 15 times higher than that of the wild-type enzyme. Although L88F and R61A showed less significant changes in KM values when compared with wild-type PETase, the kcat values of these two mutants were nearly 50% higher than that of wild-type PETase.
Since I179F showed a very significant advantage in enzymatic activity, we used scanning electron microscopy (SEM) to observe the changes in the morphology of the PET film surface (Fig. 4). When I179F was reacted with the PET film at 30°C for 48 h, the surface of the PET film underwent a significant morphological change compared with the surface that was reacted with the negative control; the surface of the PET film was obviously roughened and eroded, and a large number of holes were observed, along with a great deal of shedding.
《Fig.4》

Fig. 4. SEM of samples treated with I179F at 30°C for 48 h (2500× magnification). (a) Control sample; (b) I179F-treated sample.
《4. Discussion》
4. Discussion
Through cell-free protein synthesis, we successfully screened three variants with increased enzymatic activity. We attempted to understand the mechanism of the increase in enzymatic activity through the docking of the PETase and 2PET molecules. We analyzed the results of R61, L88, and I179 separately (Fig. 5).
《Fig.5》
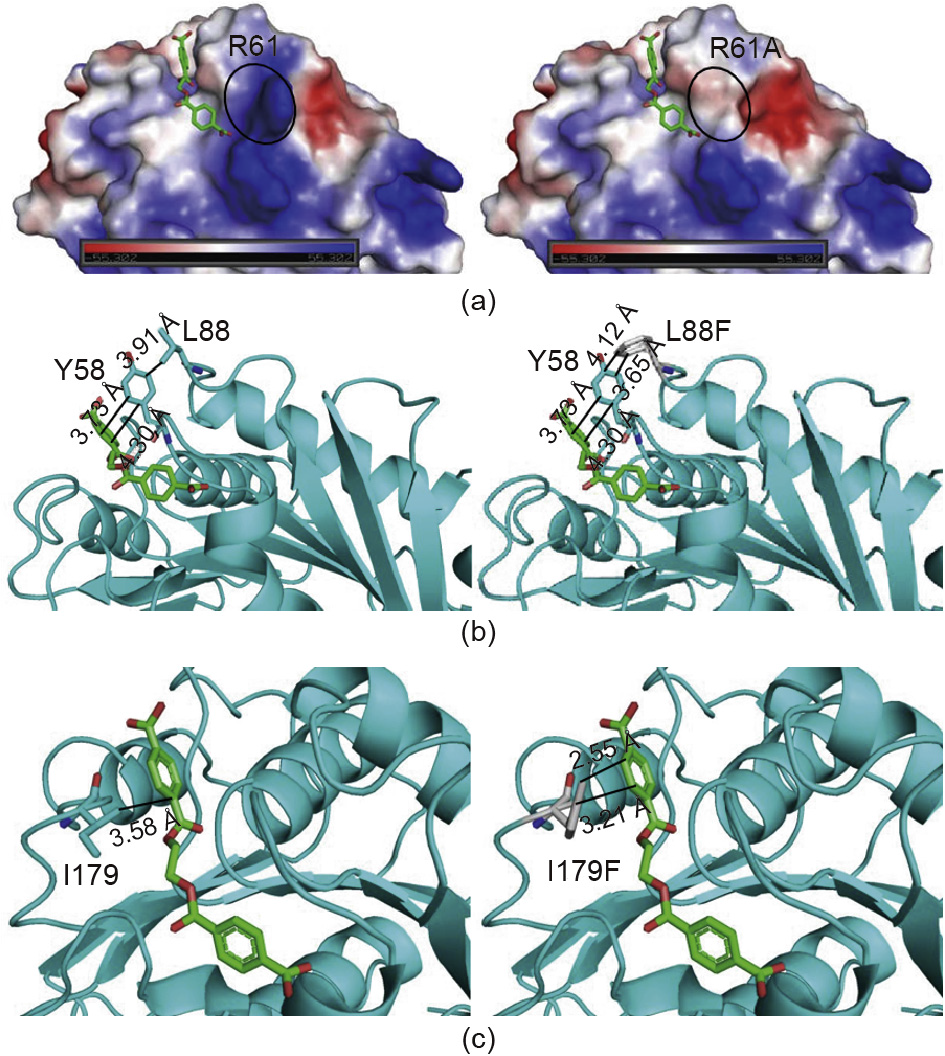
Fig. 5. Comparison and analysis of different mutant PETases. (a) Analysis of the electrostatic surface potential of wild-type R61 and mutant R61A (red, blue, and white represent negative, positive, and neutral values, respectively); (b) the structure of wild-type L88 and mutant L88F in a complex with docked 2PET in the binding side; (c) the structure of wild-type I179 and mutant I179F in a complex with docked 2PET in the binding site. (2PET shows as red and green sticks.)
R61 is located at the end of the substrate-binding groove. Variant R61A changes the surface charge around the substrate-binding groove, making the groove more hydrophobic and making it easier for the substrate to accumulate near the substrate-binding cleft (Fig. 5(a)). This explains why the kcat of R61A is 1.7 times higher than that of wild-type PETase (Table 1). On the one hand, variant R61A might eliminate the steric hindrance of the substraterelease channel. On the other hand, R61 did not interact with the substrate, so mutation would not change the enzyme-substrate interaction, as shown by the lack of change in the KM value of R61A (Table 1).
L88F stabilizes the state of Y58, which plays an important role in the substrate-binding groove of PETase [28]. The interaction between Y58 and the substrate is enhanced because L88F acts on Y58 (Fig. 5(b)). Therefore, the KM of L88F is lower than that of wild-type PETase. At the same time, since the exchange that was made in L88F enhances the interaction between the enzyme and the substrate, the catalytic PET-degradation rate of the enzyme reached 17.5 mg per μmol·L-1 PETase per day, which is 2.1 times greater than that of wild-type PETase (Fig. 3).
Through an analysis of the complex structural model of PETase and 2PET, a direct mutual hydrophobic interaction between I179 in the substrate-binding groove and 2PET was found (Fig. 1). This interaction is attributed to the binding between the enzyme and substrate molecules (Fig. 5(c)). In a previous study, when I179 was mutated to alanine, enzymatic activity was almost lost, since this interaction was attenuated [28]. In our study, in order to increase the enzymatic activity, I179 was mutated to a more hydrophobic phenylalanine residue with larger branch chains. Such mutants were designed to construct a novel PETase analog with an amino acid residue that can immobilize the phenyl ring of PET. This amino acid might function somewhat like the two inherent substrate phenyl ring-binding residues (W156/W130) that have been reported in PETase structural studies [28,35]. I179F exhibited a strong effect on the localization of the substrate and remarkably improved the catalytic efficiency. According to an in vitro enzyme reaction assay, the KM of the I179F mutant was reduced by 3.8 times and its kcat/KM value was increased by 15.6 times, compared with those of wild-type PETase (Table 1). Thus, this mutant shows the highest increase in PETase activity reported to date. To further investigate and improve the catalytic efficiency of PETase, our research will focus on multipoint mutations and saturation mutations.
《Acknowledgements》
Acknowledgements
This work was funded by National Program on Key Basic Research Project by the Ministry of Science and Technology of China (2014CB745100), the National Natural Science Foundation of China (21676190 and 21621004), and the Innovative Talents and Platform Program of Tianjin (16PTGCCX00140 and 16PTSYJC00050).
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Yuan Ma, Mingdong Yao, Bingzhi Li, Mingzhu Ding, Bo He, Si Chen, Xiao Zhou, and Yingjin Yuan declare that they have no conflict of interest or financial conflicts to disclose.
《Appendix A. Supplementary data》
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2018.09.007.











 京公网安备 11010502051620号
京公网安备 11010502051620号




