

《1. Introduction》
1. Introduction
While worldwide energy consumption continues to grow, unfortunately, nearly 88% of the current energy economy relies on fossil fuels [1]. It is only a matter of time before fossil fuels become either completely depleted or unprofitable to retrieve. Despite their outsized share of the energy portfolio, the era of fossil fuels is coming to an end [2]. Apart from the problem of diminishing availability, the negative externalities of fossil fuels have posed a prominent risk to the global ecosystem. At present, the world’s main source of energy is the combustion of fossil fuels. The byproducts of this combustion (e.g., CO2, NOx , SOx , and fine particles) seriously pollute the air, soil, and water [3–5]. It is urgent that we adopt a fresh mindset in order to find solutions to these problems and to devise a future with a more secure and sustainable energy supply. Renewable energy will play a vital role in the world’s energy future. However, there is a market barrier that stems from a major difference between renewable and conventional energy sources [6–9]. The amount of energy (mainly in the form of electricity) yielded by renewable energy sources can change unpredictably over a short period of time. For example, solar systems only produce energy when the sun is shining. Unfortunately, other renewable sources such as wind and tidal movements also possess adverse inconstancy [10]. This inconstancy makes the current generation of renewable energy less reliable than fossil-fuel-derived energy, because its output is highly dependent on weather conditions (i.e., clouds or wind) and time (i.e., day or night). For renewable energy to be practical on a very large scale, highly efficient electricity conversion and the high-density storage of electricity are required to enable energy-distribution technologies.
Electrochemical hydrogen–water conversion (H2 + O2  H2O) is a clean and efficient execute solution for a sustainable energy system [11,12]. To be specific, renewable energy can be converted into chemical energy stored in hydrogen (H2) through water electrolysis [13]. Inversely, hydrogen molecules can be electrochemically recombined into water (H2O) in order to output electricity through fuel cells. In this energy system, hydrogen acts as the energy carrier, and the energy conversion is independent of thermal cycles [1,14,15]. Because it is based on electrochemical reactions, hydrogen–water conversion can drastically reduce the release of climate-changing gases and compounds that are harmful to the natural environment and to human health. However, for the purposes of practical application, hydrogen must first be obtained, then be stored, and finally be converted back to water to release the stored energy [16]. In order to achieve this target, efficient, low-cost water electrolysis and fuel cell technology must be efficiently integrated together. Electrochemical processes are the core of these energy conversion technologies, including the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER) in the water electrolyzer, and the oxygen reduction reaction (ORR) and the hydrogen oxidation reaction (HOR) in the hydrogen– oxygen fuel cell [17,18]. The output performance of the aforementioned energy conversion technologies is significantly influenced by the efficiency of these four electrochemical reactions. Thus, the most critical problem in this sustainable energy system is how to effectively catalyze these reactions on the catalytic electrode surface in order to attain the lowest overpotential and highest current density [19–22]. Apart from the potential drop induced by electrochemical reactions, the electrical resistances and transport-related resistances affect the overall cell voltage of the water electrolysis and fuel cell. Accelerating the electron and proton transfer and the product emission by optimizing the electrode structure is another problem that requires extensive attention.
H2O) is a clean and efficient execute solution for a sustainable energy system [11,12]. To be specific, renewable energy can be converted into chemical energy stored in hydrogen (H2) through water electrolysis [13]. Inversely, hydrogen molecules can be electrochemically recombined into water (H2O) in order to output electricity through fuel cells. In this energy system, hydrogen acts as the energy carrier, and the energy conversion is independent of thermal cycles [1,14,15]. Because it is based on electrochemical reactions, hydrogen–water conversion can drastically reduce the release of climate-changing gases and compounds that are harmful to the natural environment and to human health. However, for the purposes of practical application, hydrogen must first be obtained, then be stored, and finally be converted back to water to release the stored energy [16]. In order to achieve this target, efficient, low-cost water electrolysis and fuel cell technology must be efficiently integrated together. Electrochemical processes are the core of these energy conversion technologies, including the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER) in the water electrolyzer, and the oxygen reduction reaction (ORR) and the hydrogen oxidation reaction (HOR) in the hydrogen– oxygen fuel cell [17,18]. The output performance of the aforementioned energy conversion technologies is significantly influenced by the efficiency of these four electrochemical reactions. Thus, the most critical problem in this sustainable energy system is how to effectively catalyze these reactions on the catalytic electrode surface in order to attain the lowest overpotential and highest current density [19–22]. Apart from the potential drop induced by electrochemical reactions, the electrical resistances and transport-related resistances affect the overall cell voltage of the water electrolysis and fuel cell. Accelerating the electron and proton transfer and the product emission by optimizing the electrode structure is another problem that requires extensive attention.
This article provides a comprehensive review of recent advances toward the structural engineering of electrocatalytic catalysts for electrochemical hydrogen–water conversion. Two major issues are addressed in this review: ① the origin of energy dissipation in the electrochemical hydrogen–water conversion system; and ② the structural design of electrocatalysts for high energyconversion efficiency, driven by the combination of fundamental science and practical technology. In the second section, after a brief introduction of the electrochemical process that occurs in hydrogen–water conversion, we present a review of the energy dissipation in the two functioning technologies—that is, water electrolysis and the fuel cell—from a practical standpoint, and use classical kinetics to analyze the key barrier of the electrochemical reactions occurring on the catalyst surface. With the aid of scaling relations among reactive intermediates, we develop a framework to understand catalytic trends, which ultimately provides rational guidance toward the development of improved catalysts for a wide range of reactions. In the third section, we summarize general strategies on designing higher-performance electrocatalysts and discuss their advantages and drawbacks. Featured examples of state-of-the-art electrocatalysts that have been achieved for each reaction by structural design are presented in this part, demonstrating the successful combination of synthetic chemistry, electrocatalytic chemistry, and computational chemistry. The last section outlines the key scientific problems in the electrochemical hydrogen–water conversion system and provides further development direction for catalyst design for a renewable and clean energy system with a high energy efficiency.
《2. Fundamentals of electrocatalysis》
2. Fundamentals of electrocatalysis
《2.1. Electrochemical reactions of hydrogen–water conversion》
2.1. Electrochemical reactions of hydrogen–water conversion
As illustrated in Fig. 1, two different functioning technologies are involved in this renewable and clean energy system: water electrolysis and the fuel cell. The two electrolyzers are mainly composed of four parts: the electrolyte (e.g., H2O), the ion-exchange membrane (e.g., a Nafion membrane), the anode electrode, and the cathode electrode [23,24]. In order to accelerate water splitting, the two electrodes are always coated with a highly active and stable catalyst layer. In the water electrolyzer, electrical energy is consumed to split water into gaseous hydrogen (H2) and oxygen (O2). Taking acid water electrolysis as an example, water is oxidized to form oxygen molecules and protons at the anode, as the electrons pass through the external circuit and the protons pass through the membrane down to the cathode. Meanwhile, the protons and electrons combine at the cathode to form hydrogen molecules.
The electrochemical reaction that occurs in the fuel cell is the exact opposite of the water electrolysis process [25]. A spontaneous “cold” combustion of hydrogen and oxygen occurs in the fuel cell device, in which hydrogen acts as the fuel and oxygen acts as the oxidizer. In general, hydrogen diffuses to the anode surface by penetrating through the electrode pores. Through the catalytic action of the catalyst layer, the adsorbed hydrogen is ionized and releases an electron at the electrode. Next, the hydrogen ions that pass through the electrolyte and the electrons that pass through the external circuit all reach the cathode, recombine with oxygen molecules to form water molecules, and release electricity. The heat generated by the internal reaction and electrical resistance can be removed by applying suitable water or air cooling systems. Table 1 summarizes the half-cell reactions of water electrolysis and the fuel cell in different media. The four reactions can be grouped into two reversible reaction couples: hydrogen-involving HER and HOR with an equilibrium potential (U0) of 0 V versus reversible hydrogen electrode (vs RHE); and oxygen-involving ORR and OER with a U0 of 1.23 V vs RHE. From a chemical point of view, hydrogen–water conversion is composed of two redox couples: The water/oxygen couple at high potential and the water/hydrogen couple at relatively low potential [26]. In acidic media, hydrated protons transfer charges from the anode to the cathode, while hydroxide ions act as charge carriers from the cathode to the anode in alkaline electrolytes.
《Fig. 1》

Fig. 1. Schematic illustration of water electrolysis and the fuel cell reactions in hydrogen–water electrochemical conversion.
《Table 1》
Table 1 Summary of the half-cell reactions of water splitting and the fuel cell in acid and alkaline electrolyte.

《2.2. Energy dissipation of water electrolysis and the fuel cell》
2.2. Energy dissipation of water electrolysis and the fuel cell
According to the thermodynamics of hydrogen–water conversion, reversible water electrolysis and the H2–O2 fuel cell have the same electrical onset potential of 1.23 V under standard conditions. However, the actual onset potential for the two reactions is far from the standard electrical potential. Under practical operating conditions, the cell voltage of the H2–O2 fuel cell and of water electrolysis is always below 0.9 V and higher than 1.8 V, respectively, even utilizing state-of-the-art noble metals as the electrocatalysts [2,27–29]. In practice, in order to drive the electrochemical reaction process, there are a number of barriers that must be overcome, including the electrical resistance of the circuit, activation energies of the electrochemical reactions, blockage of the electrode surfaces by the product gas bubbles and water, and ionic transfer resistances across the electrolyte solution [24]. These barriers, which require a sufficient electrical energy supply, greatly reduce the energy conversion efficiency and cause the work potential to fall short of the thermodynamic potential, which is the so-called phenomenon of polarization (or overpotential, or overvoltage) [30]. Fig. 2 shows the resistances (i.e., the barriers) in a typical cell with a liquid electrolyte [31,32]. The first resistance from both ends (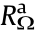 and
and 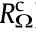 ) is the external electrical circuit resistance, which includes the internal resistance of the wiring and connections at the anode and cathode, and the resistance of the electrons across the catalyst layers, which are not always good electronic conductors [33].
) is the external electrical circuit resistance, which includes the internal resistance of the wiring and connections at the anode and cathode, and the resistance of the electrons across the catalyst layers, which are not always good electronic conductors [33]. 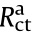 and
and  originate from the overpotential of the half-cell reactions on the surface of the anode [34].
originate from the overpotential of the half-cell reactions on the surface of the anode [34].  and
and 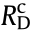 are caused by the diffusion layers close to the electrode surface when mass transport phenomena are involved or gaseous species are formed [35]. As for water electrolysis, the partial coverage of the electrode surface by generated bubbles hinders the contact between the electrode and electrolyte. Similarly, the product water acts as a blockage between the electrode and the H2 and O2 input, inducing a resistance of mass transport in an alkaline fuel cell (AFC). Rions originates from the transport of ions in the electrolyte and Rsep stems from the resistance of the cell separator [36]. A similar situation is found in other types of cells, such as zero-gap and proton-exchange membrane (PEM) cells [37,38].
are caused by the diffusion layers close to the electrode surface when mass transport phenomena are involved or gaseous species are formed [35]. As for water electrolysis, the partial coverage of the electrode surface by generated bubbles hinders the contact between the electrode and electrolyte. Similarly, the product water acts as a blockage between the electrode and the H2 and O2 input, inducing a resistance of mass transport in an alkaline fuel cell (AFC). Rions originates from the transport of ions in the electrolyte and Rsep stems from the resistance of the cell separator [36]. A similar situation is found in other types of cells, such as zero-gap and proton-exchange membrane (PEM) cells [37,38].
《Fig. 2》
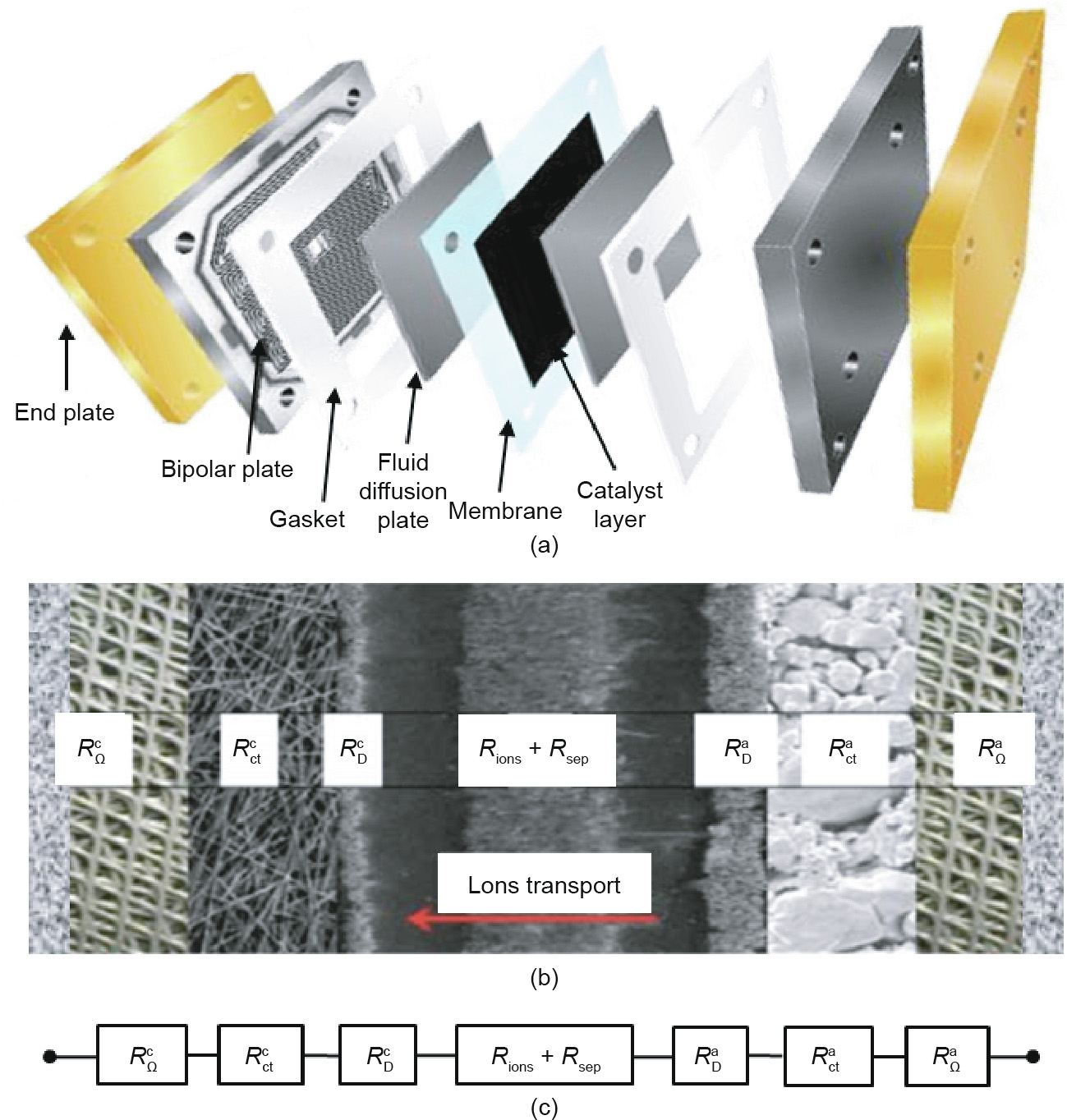
Fig. 2. (a) Schematic of a typical AFC with a liquid electrolyte; (b) cross-section of the electrolyzer; (c) equivalent electric circuit. Reproduced from Ref. [31] with the permission of Elsevier, ©2014.
The resistances in cell systems can be classified into three categories: activation resistances (losses due to electrochemical reactions), ohmic resistances (losses due to ionic and electronic conduction), and concentration resistances (losses due to mass transport). Each of the three major losses contributes to the characteristic shape of the current–voltage (i–V ) curve of the electrochemical cell [39,40]. Fig. 3 shows a typical i–V curve for water electrolysis and for a fuel cell. In the case of water electrolysis (Fig. 3(a)), the current starts to flow across the cell above the thermodynamic electrolysis voltage of 1.23 V. Additional voltage is required to overcome the resistances discussed above. At low current densities, the voltage drops caused by ohmic resistances are small, and the reaction activation overvoltage accounts for the dominant part of the voltage drop. The logarithmic shape of the polarization curve (the Tafel area) is attributed to the charge transfer phenomena at the anode and cathode. As the overvoltage increases further, the reaction activation barrier decreases, and the shape of the polarization curve becomes linear. This linear shape indicates that the ohmic resistance is now the key kinetic parameter of the cell. In the case of the fuel cell (Fig. 3(b)), the activation resistances mostly affect the initial part of the curve, the ohmic resistances are mainly apparent in the middle section of the curve, and the concentration resistances are significant in the tail. Although the reactions that occur in the two functioning technologies are reversible, the shapes of the i–V curves are not the same: the i–V curve for water electrolysis generally obeys the Butler–Volmer model even at very high overpotentials, while the i–V curve for a fuel cell tends to show a constant value at high overpotentials due to the limitation in the mass transfer rate.
《Fig. 3》

Fig. 3. Schematics of the polarization curves for (a) water electrolysis and (b) a fuel cell.
To improve the energy efficiency of the two electrochemical cells and thus improve the performance of the energy system, an understanding of these resistances must be grasped in order to minimize them. Ohmic losses are caused by the electrode material’s resistance to the electron flow and the electrolyte’s resistance to the ion flow; these can be reduced by utilizing highly conductive materials as the wiring and electrode substrate, and by diminishing the distance between the two electrodes [41–43]. The concentration losses, which are attributed to mass transport, can be relieved by increasing the pressure of the gaseous reactants or the concentration of the liquid electrolyte [44,45]. The two kinds of voltage drops mainly depend on the cell design and operation conditions. In addition to the above two resistances, the majority (> 60%) of the voltage drop in an electrochemical cell is induced by the Gibbs free-energy change for the endergonic transformation of the half-cell reactions [39]. Depending on the direction of the reactions, the activation polarizations greatly increase or decrease the anode voltage where the oxidation reaction takes place, and decrease or increase the cathode voltage where the reduction reaction occurs.
In electrochemistry, the Butler–Volmer relationship is used as the primary departure point to relate the overvoltage 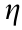 across a metal–electrolyte interface to the current density j (in A·cm-2 ) across this interface [46]:
across a metal–electrolyte interface to the current density j (in A·cm-2 ) across this interface [46]:

where is the overvoltage—that is, the difference between the actual voltage across the interface and the equilibrium voltage; j0 is the exchange current density in A·cm-2 ; a is the coefficient of the charge transfer; n is the number of electrons transferred in the electrochemical reaction; F ≈ 96 485 C·mol-1 is the Faraday constant; R is the constant of a perfect gas (0.082 J·(K·mol)-1); and T is the absolute temperature in K. The Butler–Volmer equation basically reveals that the current produced by an electrochemical reaction increases exponentially with the activation overvoltage and exchange current density. In fact, improving the reaction energy efficiency focuses on increasing j0, which represents the ‘‘rate of exchange” between the reactant and product at equilibrium. Taking the forward reaction for simplicity and including the concentration effects, j0 is defined as follows:

where c is the reactant surface concentration, 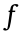 is the decay rate to products, and
is the decay rate to products, and  is the activation energy barrier for the forward reaction. Eq. (2) clearly shows that decreasing the size of the activation energy barrier () will increase j0 under a given environmental condition. In the actual reaction, only species in the activated state can undergo the transition from reactant to product. In fact, the activation energy of the reactions is strongly influenced by the electrode material [47]. A catalytic electrode is a site for species activation and transition. Using a highly catalytic electrode can significantly lower the activation barrier for the reaction, and therefore provides a way to dramatically increase j0. To reduce the activation energy of the electrode reactions, continuing research efforts are focusing on the design of efficient catalytic electrode materials based on an understanding of the relationship between the activation energies, electrode materials, and surface configurations.
is the activation energy barrier for the forward reaction. Eq. (2) clearly shows that decreasing the size of the activation energy barrier () will increase j0 under a given environmental condition. In the actual reaction, only species in the activated state can undergo the transition from reactant to product. In fact, the activation energy of the reactions is strongly influenced by the electrode material [47]. A catalytic electrode is a site for species activation and transition. Using a highly catalytic electrode can significantly lower the activation barrier for the reaction, and therefore provides a way to dramatically increase j0. To reduce the activation energy of the electrode reactions, continuing research efforts are focusing on the design of efficient catalytic electrode materials based on an understanding of the relationship between the activation energies, electrode materials, and surface configurations.
《2.3. A guide for electrocatalyst design for hydrogen–water conversion》
2.3. A guide for electrocatalyst design for hydrogen–water conversion
In terms of the reaction mechanism, the activation energy barrier () can be quantified by the Gibbs free-energy change ( ) for the rate-determining step (RDS) at the equilibrium potential, and its theoretical value on different catalytic materials can be calculated by means of density functional theory (DFT) calculation. In this way, the relationship between the activation energy and the electrode material is built, as a volcano-shaped plot is obtained by plotting j0 versus . The most common shape of the volcano plot is the HER rate description based on the Langmuir type of adsorption with the maximum located near the position where the hydrogen adsorption free energy (
) for the rate-determining step (RDS) at the equilibrium potential, and its theoretical value on different catalytic materials can be calculated by means of density functional theory (DFT) calculation. In this way, the relationship between the activation energy and the electrode material is built, as a volcano-shaped plot is obtained by plotting j0 versus . The most common shape of the volcano plot is the HER rate description based on the Langmuir type of adsorption with the maximum located near the position where the hydrogen adsorption free energy ( ) is zero [48]. In the HER, the reaction species is first adsorbed on the catalyst surface to form the reaction intermediate (M–Hads). After the aforementioned Volmer step, hydrogen molecules can be formed by the coupling of an electron and a proton in the electrolyte through a Heyrovsky step, or their direct combination via a Tafel step [30,49,50]. As a result, the is the overall decisive rate for HER [51,52]. In recent years, the DFT-calculated has been widely used as the activity descriptor [53–55], for many traditional metals, metal composites/metal alloys, and nonmetallic materials. As shown in Fig. 4(a), different metals show significant differences in HER exchange current density, and the highly active metals (e.g., Pt) located near the top of the volcano plot possess optimal [56]. If the catalytic material has a weak adsorption force on hydrogen, the hydrogen atom can barely be absorbed on the surface of the material, and the overall reaction rate is determined by the adsorption step of hydrogen (Volmer step). On the other hand, a too-strong adsorption of hydrogen atoms onto catalytic materials results in difficulty breaking the M–Hads bond to form H2, and the RDS is the desorption step (Heyrovsky/Tafel). As the reverse process of HER, the RDS of HOR is the dissociative adsorption of H2 on the catalyst surface, which involves electron transfer from the surface to the
) is zero [48]. In the HER, the reaction species is first adsorbed on the catalyst surface to form the reaction intermediate (M–Hads). After the aforementioned Volmer step, hydrogen molecules can be formed by the coupling of an electron and a proton in the electrolyte through a Heyrovsky step, or their direct combination via a Tafel step [30,49,50]. As a result, the is the overall decisive rate for HER [51,52]. In recent years, the DFT-calculated has been widely used as the activity descriptor [53–55], for many traditional metals, metal composites/metal alloys, and nonmetallic materials. As shown in Fig. 4(a), different metals show significant differences in HER exchange current density, and the highly active metals (e.g., Pt) located near the top of the volcano plot possess optimal [56]. If the catalytic material has a weak adsorption force on hydrogen, the hydrogen atom can barely be absorbed on the surface of the material, and the overall reaction rate is determined by the adsorption step of hydrogen (Volmer step). On the other hand, a too-strong adsorption of hydrogen atoms onto catalytic materials results in difficulty breaking the M–Hads bond to form H2, and the RDS is the desorption step (Heyrovsky/Tafel). As the reverse process of HER, the RDS of HOR is the dissociative adsorption of H2 on the catalyst surface, which involves electron transfer from the surface to the  antibonding orbital of the H2 molecule [57]. Consequently, the interaction of M–Hads also plays a dominant role in the kinetics of HOR, and the activity of HOR follows the same trend as HER on noble metal surfaces due to the high reversibility of these two reactions (Fig. 4(b)) [58–61].
antibonding orbital of the H2 molecule [57]. Consequently, the interaction of M–Hads also plays a dominant role in the kinetics of HOR, and the activity of HOR follows the same trend as HER on noble metal surfaces due to the high reversibility of these two reactions (Fig. 4(b)) [58–61].
In addition to the hydrogen-involved reactions, the relationship between j0 and can be applied to the oxygen-related reactions that occur in hydrogen–water conversion. As shown in Figs. 4(c) and (d), the shapes of volcano plots for these reactions are quite similar, except for the reaction intermediates determining the reaction rate. In general, the ORR includes either a fourelectron pathway to reduce oxygen to water, which is attractive for fuel cells, or a two-electron pathway, which is desirable for producing hydrogen peroxide [62]. In fact, a direct four-electron mechanism can either be a dissociative or associative process, depending on the oxygen dissociation barrier on the catalyst surface [63]. As a result, the oxygen adsorption strength ( ) is associated with the ORR activity to construct a volcano plot (Fig. 4(c)) [63,64]. For metals that bind oxygen too strongly, the reaction rate is limited by the removal of O* or OH* species. For metals that bind oxygen too weakly, the reaction rate is limited by splitting of the O–O bond in O2 (dissociative mechanism) or, more likely, by the transfer of electrons and protons to the adsorbed O2 (associative mechanism), depending on the applied potential [63]. As indicated by the volcano plot in Fig. 4(c), there seems to be some room for improvement, as even platinum (Pt) is not at the absolute peak. Metals with a somewhat lower oxygen-binding energy than Pt should have a higher ORR activity. Based on the above thermodynamic volcano plot, Viswanathan et al. [65] and Hansen et al. [66] developed a microkinetic model for ORR given that the OH binding energy is varying. They found a kinetic activity volcano that is in close agreement with the thermodynamic activity volcano, and identified an activity optimum at a 0.1 eV weaker O* binding than Pt(111) for the reduction of O2 to H2O through a four-electron pathway.
) is associated with the ORR activity to construct a volcano plot (Fig. 4(c)) [63,64]. For metals that bind oxygen too strongly, the reaction rate is limited by the removal of O* or OH* species. For metals that bind oxygen too weakly, the reaction rate is limited by splitting of the O–O bond in O2 (dissociative mechanism) or, more likely, by the transfer of electrons and protons to the adsorbed O2 (associative mechanism), depending on the applied potential [63]. As indicated by the volcano plot in Fig. 4(c), there seems to be some room for improvement, as even platinum (Pt) is not at the absolute peak. Metals with a somewhat lower oxygen-binding energy than Pt should have a higher ORR activity. Based on the above thermodynamic volcano plot, Viswanathan et al. [65] and Hansen et al. [66] developed a microkinetic model for ORR given that the OH binding energy is varying. They found a kinetic activity volcano that is in close agreement with the thermodynamic activity volcano, and identified an activity optimum at a 0.1 eV weaker O* binding than Pt(111) for the reduction of O2 to H2O through a four-electron pathway.
《Fig. 4》
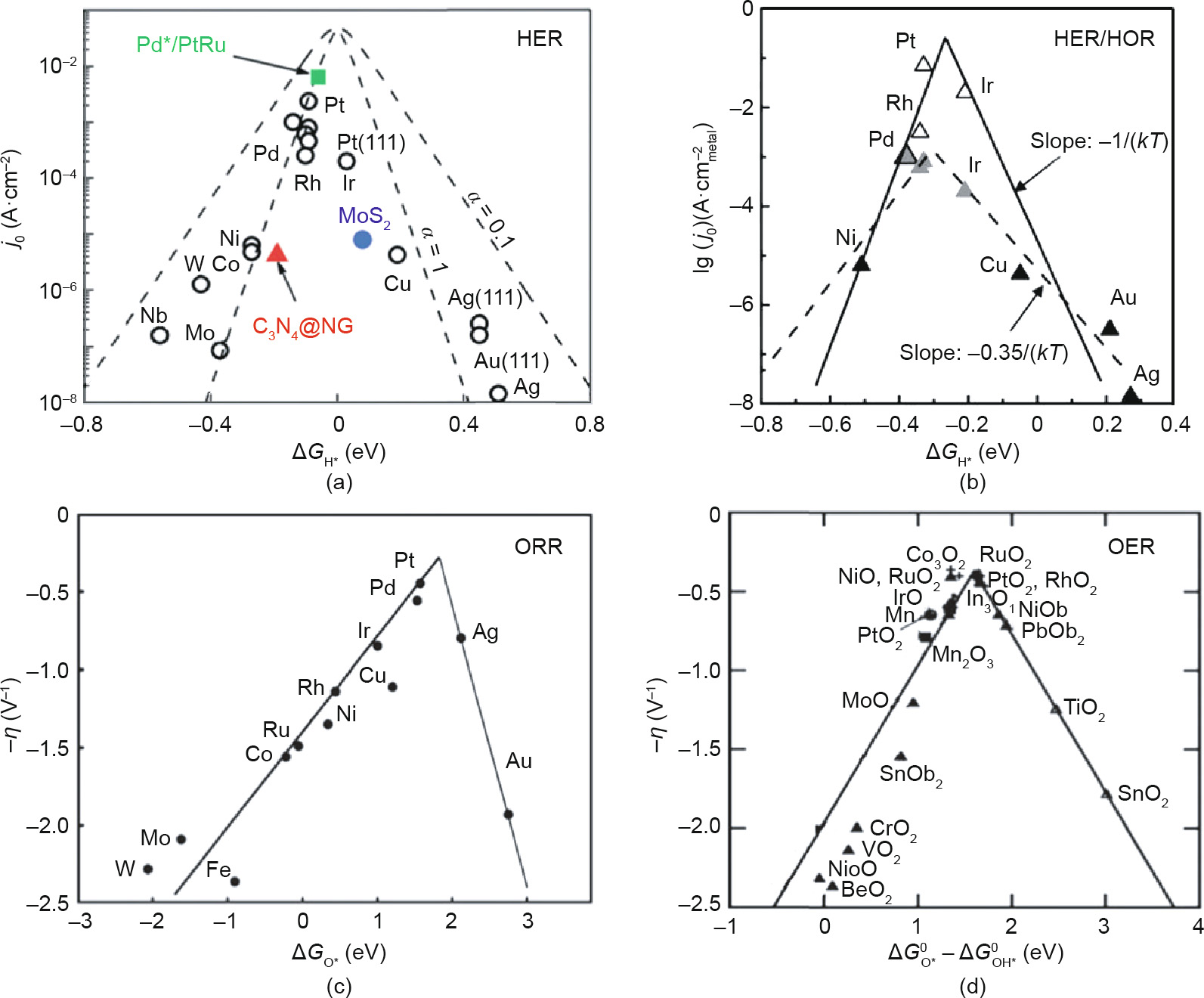
Fig. 4. (a) HER exchange current densities (j0) as a function of for the surfaces of various materials; (b) volcano plot computing the as a function of the surface normalized HER/HOR exchange current densities (j0) measured in acid electrolytes; (c) ORR activity for various metals vs ; (d) OER activity (overpotential at a certain value of current density) plotted against -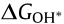 for oxides. NG: nitrogen doped graphene. (a) Reproduced from Ref. [56] with permission of ACS Publications, ©2012; (b) reproduced from Ref. [61] with permission of IOP Publishing, ©2014; (c) reproduced from Ref. [63] with permission of ACS Publications, ©2004; (d) reproduced from Ref. [70] with permission of WILEY-VCH Verlag GmbH & Co. KGaA, ©2004.
for oxides. NG: nitrogen doped graphene. (a) Reproduced from Ref. [56] with permission of ACS Publications, ©2012; (b) reproduced from Ref. [61] with permission of IOP Publishing, ©2014; (c) reproduced from Ref. [63] with permission of ACS Publications, ©2004; (d) reproduced from Ref. [70] with permission of WILEY-VCH Verlag GmbH & Co. KGaA, ©2004.
The OER volcano plot has a long history starting in 1984, when Trasatti used the transition enthalpy from the lower to higher oxidation state of metal in metal oxides as a descriptor for the OER electrocatalytic activity of oxide electrodes [67]. That pioneering work viewed the OER process as a transition between two different configurations of the surface coordination complex. Accordingly, all metal oxides that are difficult or easy to oxidize are not very active for the OER. Difficult oxidization means that the intermediates are weakly adsorbed; therefore, water dissociation is the RDS. On the other hand, easy oxidization indicates that the intermediates are strongly adsorbed, and the removal of the O* or OH* species is the RDS. In this case, the OER reactivity has been related to the oxygen adsorption free energy 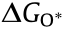 [68,69], as in the case of ORR. However, the single descriptor of for OER activity is incomplete, as the four-electron OER involves multiple intermediates (OOH*, OH*, and O*), the binding energies of which are strongly correlated and can hardly be decoupled [63,70]. A linear scaling relation exists between the binding energies of the different surface intermediates [70]; that is, if the energy associated with one reaction step is changed, the energies of the others also change. Thus, Man et al. [70] took the difference between the energy states of two subsequent intermediates (
[68,69], as in the case of ORR. However, the single descriptor of for OER activity is incomplete, as the four-electron OER involves multiple intermediates (OOH*, OH*, and O*), the binding energies of which are strongly correlated and can hardly be decoupled [63,70]. A linear scaling relation exists between the binding energies of the different surface intermediates [70]; that is, if the energy associated with one reaction step is changed, the energies of the others also change. Thus, Man et al. [70] took the difference between the energy states of two subsequent intermediates ( ) as a descriptor for the catalytic activity of several compounds, including rutile, perovskite, spinel, rock salt, and bixbyite oxides (Fig. 4(d)), whose activity obeys the volcano plot quite well. In fact, the binding energies of OH* and OOH* (whether in OER or ORR) are related to each other by a constant energy value of approximately 3.2 eV in broad classes of metal oxide materials, regardless of the binding site [70,71]. As a result of this non-ideal scaling between OOH* and OH*, a real catalyst generally shows a minimum theoretical overpotential of 0.3–0.4 V [63,72,73], even for materials at the top of the OER and ORR volcano plots, including the extensively studied RuO2 for OER [70] and Pt-based catalysts for ORR [74].
) as a descriptor for the catalytic activity of several compounds, including rutile, perovskite, spinel, rock salt, and bixbyite oxides (Fig. 4(d)), whose activity obeys the volcano plot quite well. In fact, the binding energies of OH* and OOH* (whether in OER or ORR) are related to each other by a constant energy value of approximately 3.2 eV in broad classes of metal oxide materials, regardless of the binding site [70,71]. As a result of this non-ideal scaling between OOH* and OH*, a real catalyst generally shows a minimum theoretical overpotential of 0.3–0.4 V [63,72,73], even for materials at the top of the OER and ORR volcano plots, including the extensively studied RuO2 for OER [70] and Pt-based catalysts for ORR [74].
It is noticeable that the volcano plot appropriately demonstrates the Sabatier principle [75]; that is, an ideal catalyst should bind the reaction intermediates neither too weakly nor too strongly. In other words, optimal catalytic activity can be achieved using a catalyst surface with appropriate binding energies for reactive intermediates. To be specific, the best approximation to an ideal HER/HOR catalyst would be a material that is capable of minimizing the absolute value of , and the ideal ORR and OER catalysts would be able to optimize the and , respectively. In fact, aside from decreasing the activation barrier, there is another significant way to increase j0, which is not apparent from Eq. (2): that is, to increase the number of possible reaction sites per unit area [76–79]. j0 represents the current density, or the reaction current per unit area, and the area for current density is generally based on the projected geometric area of an electrode. The true electrode surface area of an electrode with an extremely rough surface can be orders of magnitude greater than the geometric electrode area, and can thus provide many more reaction sites. Therefore, the effective j0 of a rough electrode surface will be greater than that of a smooth electrode surface. Another simple way to increase the density of active sites is to enlarge the amount of catalyst on a given electrode. However, an excessive amount of catalyst will hinder the charge and proton transfer on the electrode surface. As a result, the activity of the electrode does not increase linearly with the amount of catalyst.
In conclusion, there are two general methods for increasing the activity (or rate of reaction) of an electrocatalyst system: ① improving the intrinsic activity of each active site; and ② increasing the density of active sites on a given electrode. Both methods have pros and cons. The difference in intrinsic activity between different catalysts may be more than ten orders of magnitude, while the difference in activity caused by catalyst loading will be only 1–3 orders of magnitude. Improving the intrinsic activity of each active site is the most fundamental and effective way to achieve high activity, and its realization must be based on a deep understanding of the reaction mechanism and material properties. Increasing the number of active sites is an easier strategy, but the activity growth is limited. At the same time, activity promotion by increasing catalyst loading is obtained through the sacrifice of increasing the electrode cost and the charge and proton transfer blockage. In practice, the two methods are not mutually exclusive and can ideally be implemented simultaneously, thus greatly enhancing the activity of catalyst.
《3. Materials design for electrochemical hydrogen–water conversion》
3. Materials design for electrochemical hydrogen–water conversion
《3.1. Nanoarchitecture》
3.1. Nanoarchitecture
It is well known that the current density of a catalyst increases as the density of the active sites increases. Exposing more active sites is important in achieving high catalytic performance. Nanoarchitecture has been considered to be the most effective strategy, as it allows the density of active sites to be directly enriched and utilized, and thus efficiently optimizes the electrocatalytic activity [80–84]. The relation between the actual active surface area and the overall performance of an electrocatalyst was first recognized in transition-metal alloy systems. As early as 1982, Brown et al. [85] found that an alloy surface is generally rougher than that of a single metal, and can provide more active sites for a catalytic reaction. With the aid of nanostructuring and selective etching of molybdenum (Mo) in Ni–Mo alloys [85–87], the surface area of Ni–Mo alloys greatly increased, resulting in an obvious improvement in catalytic reactivity. With the rapid development of synthesis techniques, a series of electrocatalytic nanomaterials with different morphologies have been achieved in the past decade, including nanocages, nanofibers, nanoflowers, nano-foam, nanonets, nano-needles, nano-rings, nano-shells, and nanowires [77,88–91]. Faber et al. [92] reported metallic cobalt disulfide (CoS2) as a highly active catalyst for HER, and demonstrated the crucial role of geometric structure in determining its overall catalytic performance. Compared with the common morphologies of nanoparticles and nanofilm, an increase in active surface area drastically improves the HER catalytic performance of microstructured and nanostructured electrodes (Fig. 5), endowing the CoS2 nanowire electrodes with overpotentials as low as 145 mV for driving a current density of -10 mA·cm-2 . In addition, nanostructuring possesses a dual function in terms of both operational stability and reaction rate through the facilitation of mass transport and the removal of generated gas bubbles or water from the catalyst surface. Our group synthesized Mo2C/C with a two-dimensional (2D) lamellar structure via a controllable synthesis using a selfassembly and pre-shaping strategy [93]. The highly dispersed Mo2C nanoparticles and the 2D lamellar structure effectively boosted the mass and charge transfer across the Mo2C active sites, facilitating the electrochemical HER process. Moreover, our group further synthesized a series of three-dimensional (3D) nanostructured catalytic materials, including the NiCo2(SOH)x nanoflower [94], the coral-like FeNi(OH)x [95], the Ni–VC nanoboscage [96], Ni–Mo2C nanowire [97], and the Ni(OH)2@Ni2P nanopillar [98]. All these materials possessed highly active surfaces, fast electron transfer, and gas escape channels, which are beneficial for catalyzing water electrolysis.
《Fig. 5》
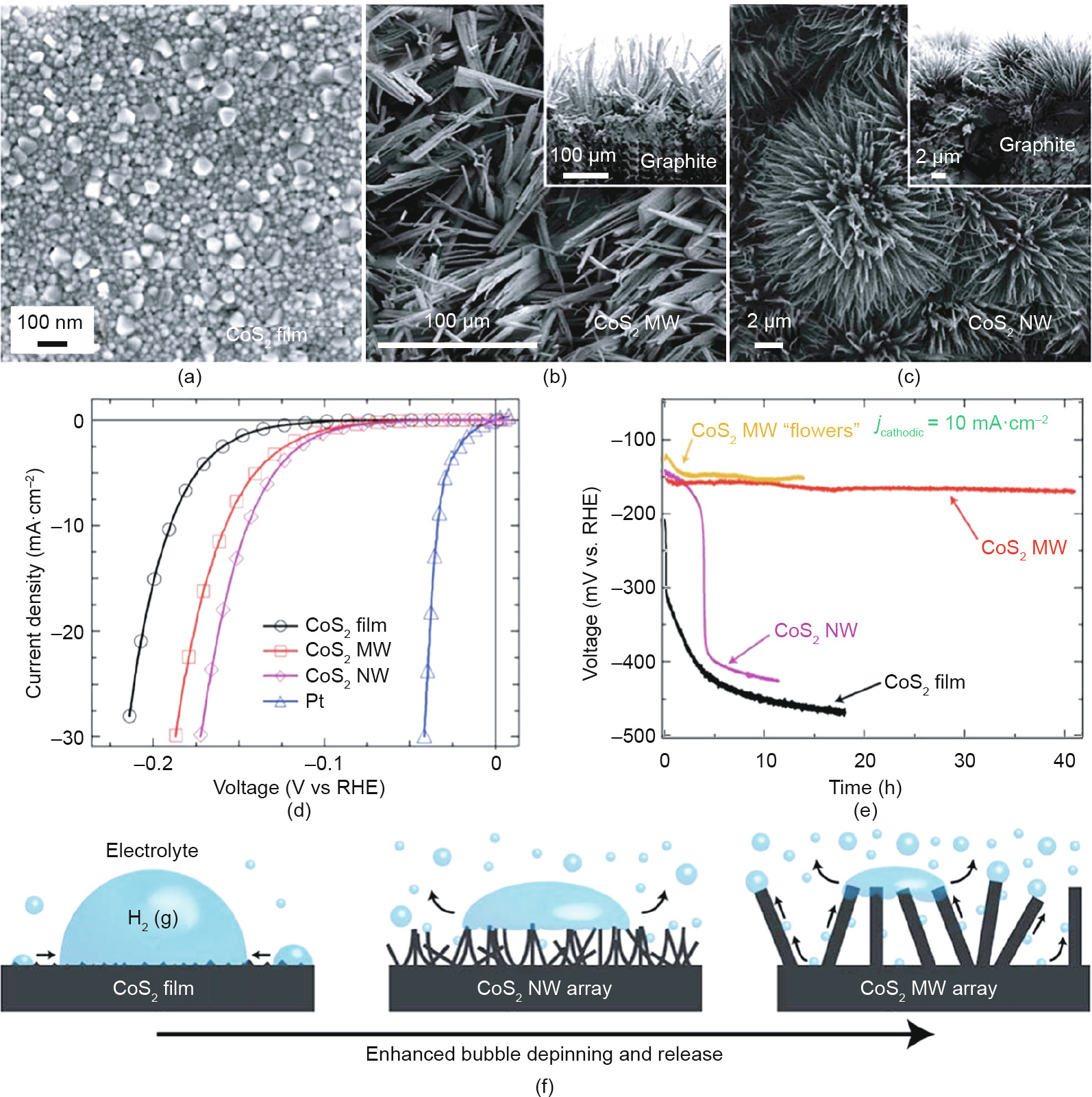
Fig. 5. (a–c) Scanning electron microscope (SEM) images of CoS2 with different morphology; (d) electrochemical characterization and (e) long-term stability measurements of CoS2 electrodes; (f) schematics of hydrogen gas bubble release from CoS2 with different structures. MW: microwire; NW: nanowire. Reproduced from Ref. [92] with permission of ACS Publications, ©2014.
To be specific, a rapid loss resulting from water flooding may take place in ORR catalysts, apart from deactivation during a long operation time [99,100]. Water flooding will interrupt the O2 supply to active sites as the porous channels are obstructed by the accumulation of water, resulting in the termination of ORR in the flooded region [101,102]. In order to quantify the mass transfer and anti-flooding performance with the pore characteristics of electrocatalysts in a fuel cell, Wang et al. [103] designed a special “rattle-drum”-like work electrode for ORR catalysts. Benefiting from a bigger pore volume and regular pore arrangement, the dual-porosity Pt/C catalyst exhibited four times the quantified mass transfer and anti-flooding efficiency of a commercial catalyst. In fact, different types of pores have special functions in the ORR process. Mesopores and macropores may be significant in mass transport during the ORR process [104–106], while micropores are beneficial for hosting most catalytic sites [107,108]. With the purpose of constructing hierarchically porous structures, the sacrificial template method has been widely adopted, using silica colloid [105,109–111], ordered mesoporous silica [106,112–114], polystyrene microspheres [115], and some other oxides [116,117] as templates. For example, a colloidal silica template was used by Liang et al. [118] for the synthesis of N-doped carbon catalyst with a high specific surface area of 1280 m2 ·g-1 , a hierarchically porous structure with meso/micropore distribution. However, the subsequent removal of the template can be time consuming and usually requires the usage of a strong acid or alkaline solution, which is dangerous to researchers and harmful to the environment. To avoid these disadvantages, our group developed a morphologycontrolled approach using NaCl as the template, in which the template can be removed using hot water and then recycled [119– 122]. A nanostructured polyaniline (PANI) with a special structure was encapsulated in the NaCl crystal via salt recrystallization, and then accurately converted into a carbon nanomaterial under high temperature (Fig. 6). Moreover, a mass of pores was created in the carbon nanomaterial by gasification in a closed nanoreactor. Due to the multiple types of pores and high utilization of active sites, the 3D Fe/N–C catalysts exhibited excellent catalytic performance toward the ORR.
《Fig. 6》
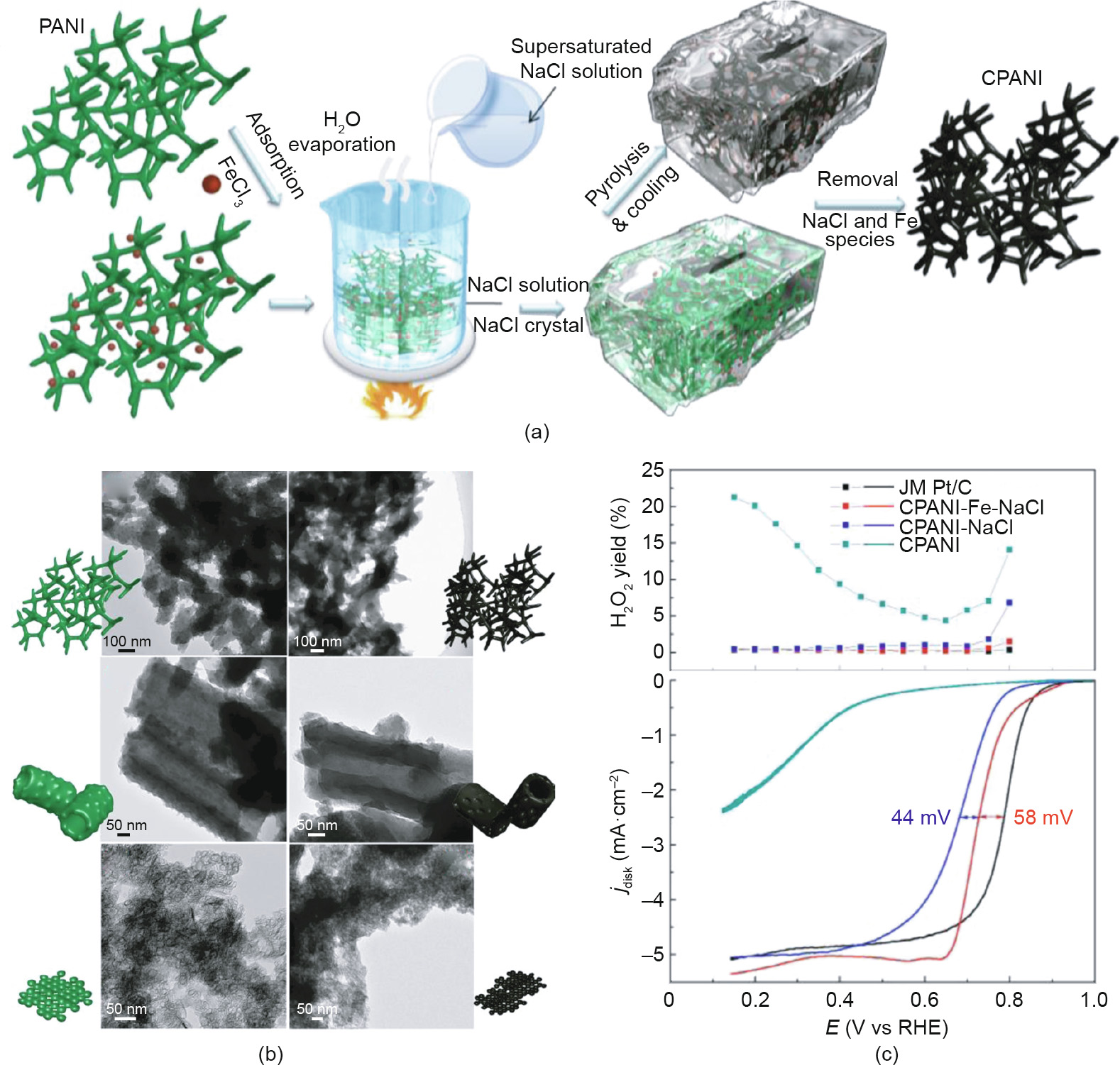
Fig. 6. (a) Schematics of the salt recrystallization method for shape fixing; (b) transmission electron microscope (TEM) images of synthesized 3D PANI and their corresponding carbonized products (CPANI); (c) H2O2 yield curves and ORR curves of different samples. JM: Johnson Matthey. Reproduced from Ref. [119] with permission of ACS Publications, ©2015.
《3.2. Facet engineering》
3.2. Facet engineering
Facet engineering is another widely studied method to modulate the catalytic performance of materials for a given reaction. The reactivity of catalytic materials is highly related to their exposed facets because the adsorption strength of the intermediate species of the catalytic reaction varies greatly on different surface facets of the catalyst. Facets are always denoted by Miller indices. The exposed facet(s) of a nanomaterial strongly correlate to the shape of the nanoparticle [123,124]. In general, faceted nanomaterials can be categorized into low-index and high-index faceted types [125]. Low-index facets are those for which the sum of the three components of the Miller indices (hkl) is small, whereas a high-index facet contains at least one Miller index greater than unity.
The structural sensitivity of the reactivity of low-index faceted nanomaterials has been demonstrated with regard to singlecrystal Pt for HER. The surface morphology of Pt(hkl ) with welldefined surfaces was confirmed by scanning tunneling microscopy (STM, Fig. 7), and the degree of activity was observed to follow the order (110) > (100) > (111) in alkaline solution [126]. Importantly, the activity in alkaline and acid electrolyte was found to differ substantially [127–129]. This pH effect involved structure–function relationships in the HER, and was further studied by Strmcnik et al. [130] with a focus on both Pt(111) and Pt(111) modified by Pt islands. Compared with the HER activity of the pristine Pt(111) surface, the HER on the Pt islands/Pt(111) electrode was found to be 5–6 times more active in alkaline solution, but only 1.5 times more active in acid electrolyte. The effect of pH on HER activity was shown to be due to the special ability of edge-step sites to dissociate water [131–135]. In addition, the activity order of Pt(hkl ) is in line with the density of low-coordinated Pt atoms due to the accelerated water dissociation step on their metal surfaces [136–139].
《Fig. 7》
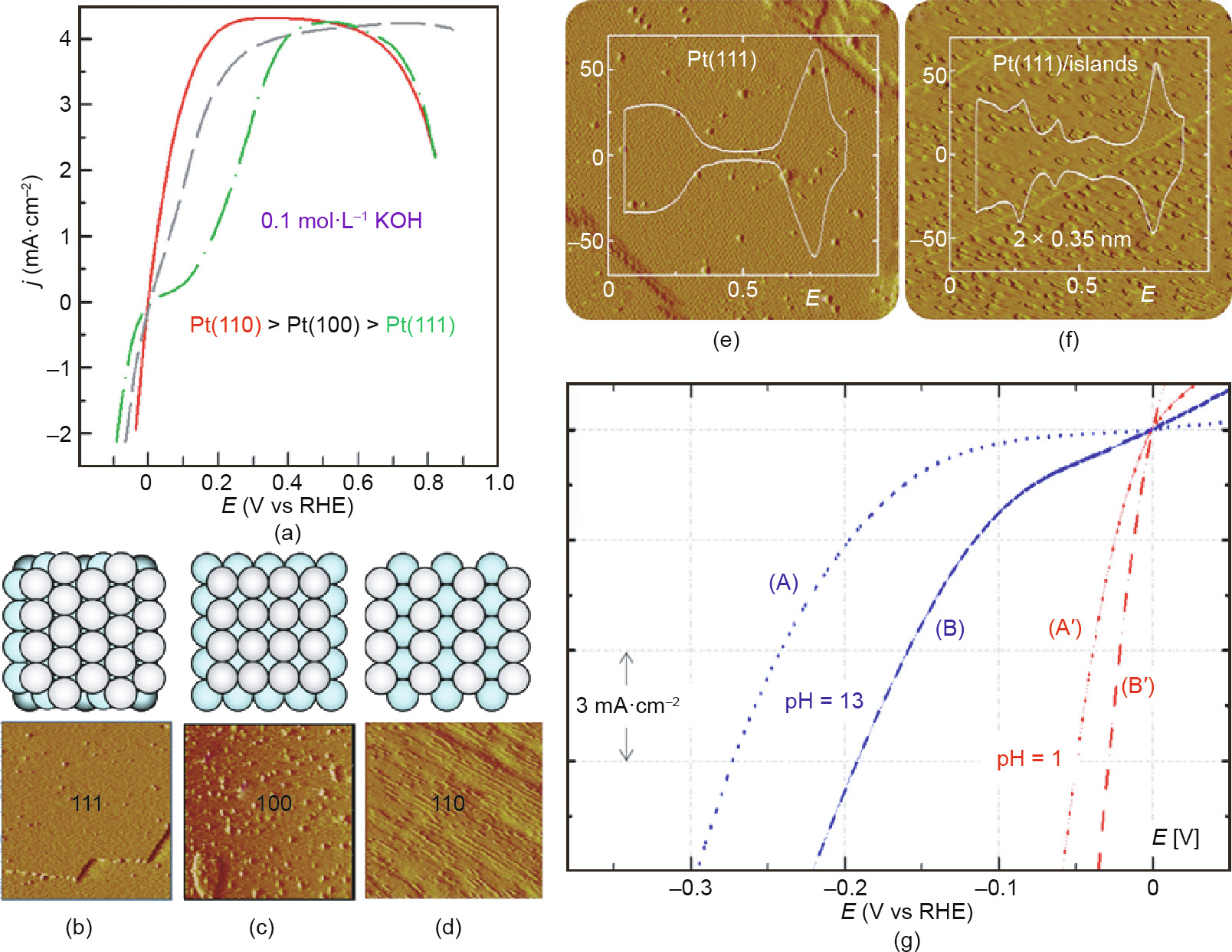
Fig. 7. (a) HER/HOR on different facet surfaces of Pt; (b–d) STM images of different facets of Pt (the insets are their corresponding structural models STM images for (e) the prepared Pt(111) and (f) the Pt islands/Pt(111) surface; (g) the HER electrochemical responses of the two Pt facets. (a–d) reproduced from Ref. [126] with permission of ACS Publications, ©2008; (e, f) reproduced from Ref. [130] with permission of Springer Nature Limited, ©2013.
A similar phenomenon was observed in the case of ORR activity on single-crystal Pt surfaces following a descending order in nonadsorbing HClO4 electrolytes [140]. However, when the electrolyte was replaced by H2SO4, Pt(100) was found to be more active than Pt(111) [141–143]. This activity difference was attributed to the special adsorption behaviors of the bisulfate anion on Pt(111). The bisulfate anions can be adsorbed on the Pt(111) surface more strongly than on Pt(100), resulting in an impeditive ORR process. Systematic investigations confirmed that the different properties of the respective facets have a significant effect on their catalytic performance. Following this work, many studies focused on developing a facet-control method to construct more active facets on a catalytic surface, from ideal single-crystal metals to more practical nanomaterials [144–150]. Narayanan and El-Sayed [144] were the first to demonstrate facet-controlled synthesis in the case of Pt nanocrystals, including (100)-terminated nanocubes, (111)- bounded nanotetrahedra, and nanospheres with both (111) and (100) facets. A high-temperature organic phase method was reported by Wang et al. [145] for the synthesis of monodispersed (100)-terminated Pt nanocubes for ORR. The facet-controlled Pt nanocubes showed a specific ORR activity that was more than double the activity of a commercial Pt catalyst in acid electrolyte.
Because of the higher density of low-coordinated atoms, steps, edges, and kinks, metals and compounds with high-index facets generally possess greatly improved reactivity in comparison with typical low-index materials [151]. However, such high-index facets are thermodynamically instable due to their higher surface energy [152,153]. Accordingly, the synthesis of high-index faceted nanomaterials has become a herculean task. In the last few years, a wide variety of synthetic protocols have been exploited for the synthesis of metallic nanomaterials with high-index facets for the purpose of improved catalytic reactivity. Yu et al. [154] developed a simple reduction route in aqueous solution to prepare Pt concave nanocubes (c-NCs) enclosed by high-index facets of (510), (720), and (830). Pt c-NCs have also been fabricated by using glycine to manipulate the reduction kinetics of H2PtCL6 [155]. Employing electrochemical means, Tian et al. [156] synthesized tetrahexahedral (THH) Pt nanocrystals with facets including (730), (210), and (520). In addition to these single-metal materials, multi-metallic high-index faceted nanocrystals have been developed [157–160]. As shown in Fig. 8, Luo et al. [159] reported a new class of Pt3Fe zigzag-like nanowires (z-NWs) with stable high-index facets and a nanosegregated Pt-skin structure. These unique structural features endowed the Pt-skin Pt3Fe z-NWs with a mass and specific ORR activity of 2.11 A·mg-1 and 4.34 mA·mg-2 , respectively, at 0.9 V vs RHE.
With the purpose of reducing catalyst cost, the design and preparation of low-cost metal nanomaterials with different exposed reactive facets have been a recent trend in facet engineering [161–167]. Su et al. [168] studied the growth mechanism of the NiO crystal and found that the surface energy of the NiO facets followed the order of (100) < (113) < (101) ≈ (110). Han et al. [161] provided a template-free hydrothermal method for the controllable fabrication of a surface-tailored Co3O4 nanocube (NC), nanotruncated octahedron (NTO), and nanopolyhedron (NP), with facets of (001), (112), (001) and (111). The different crystal planes endowed the Co3O4 nanocrystal with different exposed surface atomic configurations of the Co2+ and Co3+ active sites. The unusual (112) plane-enclosed Co3O4 nanoparticle on reduced graphene oxide (rGO) with abundant Co3+ sites exhibited superior activity for both OER and ORR. In addition to these metal oxides, many other metal compounds with special facets have been reported. Feng et al. [163] further synthesized Ni3S2 nanosheet arrays with stable (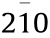 ) facets, and demonstrated them to be efficient and ultra-stable electrocatalysts for HER and OER. Wang et al. [162] obtained flower-like nickel phosphide with different crystalline structures (Ni5P4 and Ni2P) and ascribed the excellent HER activity to the hierarchical structure with high-energy (001) facets.
) facets, and demonstrated them to be efficient and ultra-stable electrocatalysts for HER and OER. Wang et al. [162] obtained flower-like nickel phosphide with different crystalline structures (Ni5P4 and Ni2P) and ascribed the excellent HER activity to the hierarchical structure with high-energy (001) facets.
《Fig. 8》
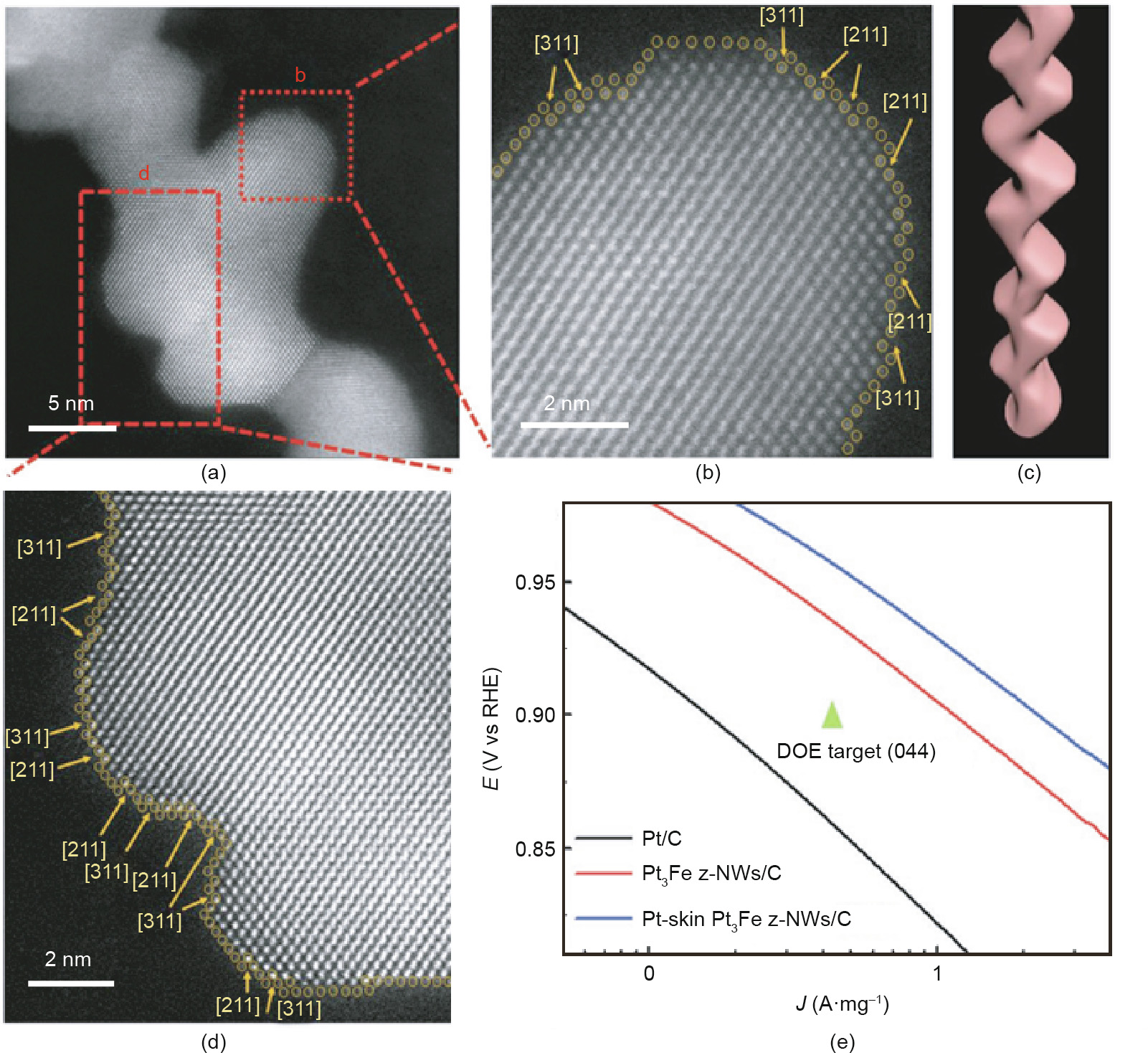
Fig. 8. (a) High-angle annular dark field (HAADF)-TEM image of Pt-skin Pt3Fe z-NWs; (b, d) enlarged images of the red square in (a); (c) structural demonstration of the z-NWs; (e) ORR mass activity Tafel plots for commercial Pt/C, Pt3Fe z-NWs/C, and Pt-skin Pt3Fe z-NWs/C. DOE: department of Energy. Reproduced from Ref. [159] with permission of WILEY-VCH Verlag GmbH & Co. KGaA, ©2018.
《3.3. Polymorph engineering》
3.3. Polymorph engineering
Apart from tuning the exposed facets of the nanocrystal, modulating the atomic scale arrangement (i.e., the transformation of the crystalline phase) can affect the intrinsic activity of a catalyst, due to the fundamental changes in its physical and chemical properties. Transition-metal disulfides with several unique polymorphs are typical cases that have been widely studied. Among these polymorphs, the metastable 1T phase has recently aroused great research interest owing to its metallic behavior, which is beneficial to electrocatalytic processes [169–174]. Lukowski et al. [169] synthesized metallic 1T-MoS2 nanosheets from semiconducting 2HMoS2 via a lithium intercalation method. The resulting 1T-MoS2 exhibited a dramatic improvement in electrocatalytic HER performance compared with the corresponding 2H polymorphs (Fig. 9). Similarly, metallic 1T tungsten disulfide (1T-WS2) was further synthesized by Lukowski et al. [173] via a simpler microwave-assisted intercalation. The polymorph engineering endowed the 1T-WS2 with faster electrical conductivity and more intensive active sites, boosting its HER activity.
《Fig. 9》

Fig. 9. (a) Phase transition of semiconducting 2H-MX2 to metallic 1T-MX2 via lithium intercalation; (b) HER polarization curves for both forms of MoS2. Reproduced from Ref. [169] with permission of ACS Publications, ©2013.
Not only can unique properties be induced by atomic arrangement, but the active sites of 1T-catalysts may also differ from those of the traditional 2H-structured phase. In this context, Voiry et al. [175] obtained highly conductive 1T-MoS2 nanosheets with excellent HER activity after removing excess negative charges from the surface of chemically exfoliated MoS2 nanosheets. Interestingly, after partial oxidation of the 1T- and 2H-MoS2, a sharp contrast in HER activity changes was observed. Although there was almost no shift in the HER activity of 1T-MoS2 after edge oxidation, the activity of 2H-MoS2 seriously decreased. It is well known that the edges of the usual 2H-MoS2 crystal are the main active sites for HER. The significant difference in HER activity between the partially oxidized 1T- and 2H-MoS2 nanosheets revealed that the main active sites of 1T-MoS2 for driving HER catalysis are not the edges of the nanosheets, but the basal planes of the nanosheets.
The catalytic performance of metal oxides may also vary with their crystal phases. Our group found that reversing spinel crystalline structure has a great influence on the ORR catalytic activity of spinel (Fig. 10) [176,177]. By adjusting the iron (Fe) content, the spinel structure of a Co–Fe-based crystal can be changed from its normal structure to the inverse structure and then back again [178]. The electrochemical results revealed that the inverse spinel {Co}[FeCo]O4/NG (nitrogen doped graphene) had the best ORR activity, outperforming commercial Pt/C. DFT results further disclosed that the higher ORR activity of the inverse-structured {Co} [FeCo]O4 could be ascribed to the modulated oxygen adsorption energy and elongated adsorbed oxygen bond induced by the dissimilarity effect of Fe and Co atoms at the octahedral site. The effect of crystal phase on the reaction pathway of ORR has also been studied. Karunagaran et al. [179] synthesized four kinds of iron oxide nanoparticles with different phases incorporated inside 3D rGO aerogels and determined their electrochemical, catalytic, and electron transfer properties for ORR. The results showed that ORR was catalyzed by all four catalysts via a two-electron pathway under higher potentials (0.70 V). On the other hand, when the potentials decrease to 0.20 V, rGO composites containing magnetite, maghemite, and goethite proceeded via four-electron transfer kinetics, whereas the hematite-containing composite went through two-electron transfer kinetics.
《Fig. 10》

Fig. 10. (a) Structure reversing and the relationships between ORR properties; (b) the ORR activities of {Co}[Fe2]O4/NG, {Co}[Co2]O4/NG, {Co}[FeCo]O4/NG, and Pt/C. Reproduced from Ref. [176] with permission of WILEY-VCH Verlag GmbH & Co. KGaA, ©2016.
Configuration distortion induced by the Jahn–Teller effect for transition-metal compounds has also been studied in relation to the electrocatalytic performances of such compounds [180]. Recently, Liu et al. [181] observed an obvious structural distortion in Co3S4 atomically thin nanosheets (CSATNs) via the ultrasound exfoliation treatment of an intermediate Co3S4/TETA hybrid precursor. The structural distortion of CSATNs generates an electronic configuration change. Compared with bulk samples, the shift from spectral to lower magnetic fields (Figs. 11(a) and (b)) implies that the spin state of Co3+ in the octahedral sites ( ) of CSATNs adjusts from low spin to high spin. High-angle annular dark field (HAADF) images showed that the octahedral coordinated cations were solely exposed in the planes, which further revealed the existence of Jahn–Teller elongation (Figs. 11(c)–(f)). Due to the synergistic adjustment in the atom and electron configuration, CSATNs possess significantly enhanced OER performance in comparison with bulk samples. In fact, the Jahn–Teller effect is attributed to the uneven electron distribution of the central ions in degenerate d orbitals (t2g or eg). Thus, the filling state of electrons in the eg orbital may have a significant role in the catalytic properties of the transition-metal compounds. A volcano relationship between the intrinsic ORR activity and the filling states of the eg orbital in the B ions of perovskite-based oxides (ABO3) was discovered by Suntivich et al. (Fig. 11(g)) [182]. The perovskite-based oxides with only one electron filling in the eg orbital (defined as eg ≈ 1) were demonstrated to possess the highest ORR activity, as O2 can adsorb on the B sites end-on with an optimal binding energy. The eg occupancy theory can be further extended to spinel oxides, although the ORR active sites of spinel are not tetrahedral sites but octahedral sites (Fig. 11(h)) [183].
) of CSATNs adjusts from low spin to high spin. High-angle annular dark field (HAADF) images showed that the octahedral coordinated cations were solely exposed in the planes, which further revealed the existence of Jahn–Teller elongation (Figs. 11(c)–(f)). Due to the synergistic adjustment in the atom and electron configuration, CSATNs possess significantly enhanced OER performance in comparison with bulk samples. In fact, the Jahn–Teller effect is attributed to the uneven electron distribution of the central ions in degenerate d orbitals (t2g or eg). Thus, the filling state of electrons in the eg orbital may have a significant role in the catalytic properties of the transition-metal compounds. A volcano relationship between the intrinsic ORR activity and the filling states of the eg orbital in the B ions of perovskite-based oxides (ABO3) was discovered by Suntivich et al. (Fig. 11(g)) [182]. The perovskite-based oxides with only one electron filling in the eg orbital (defined as eg ≈ 1) were demonstrated to possess the highest ORR activity, as O2 can adsorb on the B sites end-on with an optimal binding energy. The eg occupancy theory can be further extended to spinel oxides, although the ORR active sites of spinel are not tetrahedral sites but octahedral sites (Fig. 11(h)) [183].
《Fig. 11》

Fig. 11. (a, b) Electron paramagnetic resonance (EPR) of bulk and CSATNs; (c) HAADF image and (d) intensity line profiles of CSATNs; (e) schematic of the Jahn–Teller distortion and (f) structural transformation; (g) ORR activities of perovskite oxides as a function of the eg electrons; (h) role of eg occupancy of the active element at the octahedral site in the ORR activities of spinel oxides. (a–f) reproduced from Ref. [181] with permission of WILEY-VCH Verlag GmbH & Co. KGaA, ©2015; (g, h) reproduced from Ref. [183] with permission of WILEY-VCH Verlag GmbH & Co. KGaA, ©2017.
《3.4. Amorphization》
3.4. Amorphization
Amorphization to modulate the atomic scale arrangement, and thus increase the catalytic performance, is another research hotspot [184–189]. Short-range atomic arrangements of amorphous phases are beneficial for increasing the density of active sites [190–194]. As early as 1995, Weber et al. [190] investigated the structural units of the amorphous compound MoS3 and found that all molybdenum is present in the Mo4+ oxidation state, while sulfur atoms occur in two different types of coordination: S2– and S2 2–. Merki et al. [191] and Benck et al. [192] then confirmed that the amorphous MoS2 is more active in catalyzing HER. Structural measurements demonstrated that an amorphous MoSx film is extremely rough in surface and sulfur-rich in composition, resulting in a large active area and intensive active sites for HER catalysis. Benck et al. [192] further revealed that the increase in the HER activity of amorphous molybdenum sulfide is contributed to by the large number of active sites caused by the amorphous structure and rough, nanostructured morphology, as the activity scales with the electrochemically active surface area. Meanwhile, Li et al. [195] and Li et al. [196] systematically studied the origin of the catalytic activity of the amorphous MoS2 in terms of the composition and crystallinity. Interestingly, the experimental results revealed that the crystallinity is crucial for determining the catalytic performance, whereas the composition is not particularly significant.
In addition to the HER catalysis, Smith et al. [185] demonstrated that amorphous materials are more active than the comparable crystalline materials for OER catalysis, based on a study of the mixed-metal oxides of iron, nickel (Ni), and cobalt (Co). Due to the amorphous structure, the distribution of the metals in the amorphous films is homogeneous and their compositions can be accurately controlled. Modulated a-Fe100-y-zCoyNizOx with an optimal element content exhibited excellent catalytic property that was even comparable to that of commercial noble metal oxide catalysts. Thanks to the controllable composition of amorphous materials, the effect of metal composition on electrocatalytic performance can be further studied along with the effect of amorphization. Smith et al. [184] prepared 21 complex metal oxide films for electrocatalytic water oxidation, and demonstrated the excellent stoichiometric concentrations of Fe, Co, and Ni in each sample. Structural characterization and electrochemical measurement confirmed that iron content is important for lowering the Tafel slope, and that cobalt or nickel are beneficial in reducing the overpotential (Fig. 12). For scale-up production, Kuai et al. [186] proposed an aerosol-spray-assisted method by which amorphous mixed-metal oxides can be sustainably obtained, which is very suitable for industrial applications. The obtained Fe6Ni10Ox exhibited a low overpotential of 0.286 V for driving 10 mA·cm-2 and a small Tafel slope of 48 mV·decade-1 for the electrochemical OER, exceeding the best catalytic performance of all investigated Fe–Ni–Ox series.
《Fig. 12》

Fig. 12. Contour plots for the activity parameters of amorphous metal oxide films with different metal contents. (a) Onset potential; (b) Tafel slopes; (c) overpotential at j = 0.5 mA·cm-2 . Reproduced from Ref. [184] with permission of ACS Publications, ©2013.
Although the active sites of amorphous catalysts can be greatly enhanced by amorphous engineering, the electrical conductivity of the amorphous materials will be decreased due to short-range disorder in the crystal structure. Coupling these low-conductive materials with highly conductive materials is an effective route to guarantee the excellent electrocatalytic performance of amorphous catalysts. For example, Lee et al. [197] synthesized amorphous MnOx nanowires supported by Ketjenblack (KB) carbon as highly efficient ORR electrodes. The low-cost and highly conductive KB acts as a supporting matrix for the catalyst, greatly accelerating the electron transfer during the electrocatalytic processes. Many other amorphous/conductive composite materials, such as amorphous MoSx /carbon composite catalyst [198], amorphous MoSx /polypyrrole copolymer film (PPy/MoSx) [199], and amorphous MoSx /N-doped CNT (NCNT) forest hybrid catalyst [200], have also been reported. The highly conductive skeletons in these composite materials can overcome the barriers induced by the low electrical conductivity of amorphous catalysts, leading to a remarkable increase in catalytic activity (Fig. 13). Porous metal nanostructures, such as Ni foam [201] and nanoporous gold [202], are also used as conductive substrates to support an amorphous MoSx catalyst, of which the HER activity can be significantly enhanced.
《Fig. 13》
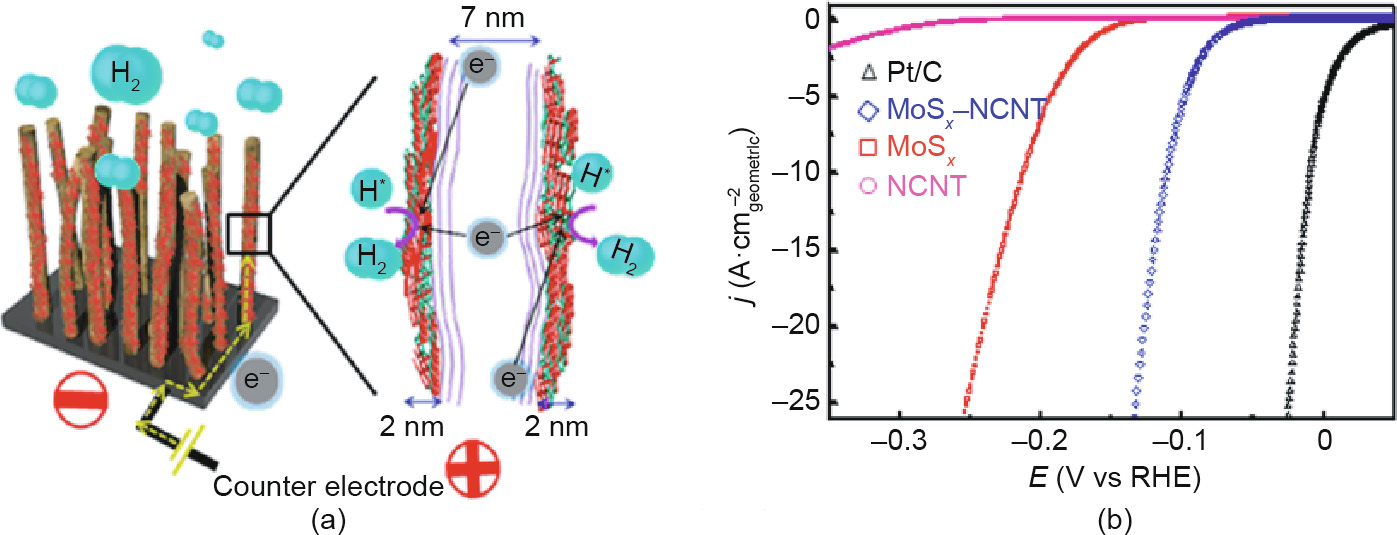
Fig. 13. Electrochemical catalytic activities. (a) HER scheme for MoSx/NCNT forest hybrid catalyst; (b) HER activities of different samples. Reproduced from Ref. [200] with permission of ACS Publications, ©2014.
《3.5. Defect engineering》
3.5. Defect engineering
Defects exist widely in nanomaterials. It has been realized that the surface of catalysts with defects always exhibit higher reactivity than the defect-free sites [203–206]. Accordingly, defect engineering has gradually developed as an effective method to tune the electronic and surface properties of nanomaterials [207–209]. Cheng et al. [210] synthesized tetragonal or cubic Mx Mn3–x O4 spinels by reducing the amorphous MnO2 in aqueous M2+ solution under ambient conditions. Due to its highly active area and abundant defects, nanocrystalline Cox Mn3–x O4 is endowed with considerable catalytic activity for both ORR and OER. Similarly, Ma et al. [211] synthesized oxygen vacancy (OV) defect-rich mesoporous MnCo2O4 materials, and found that their stability and methanol tolerance ability even exceeded those of a Pt/C catalyst. In order to obtain insight into the effect of defects in catalytic performance, our group performed a DFT + U calculation of OV concentration on the electronic structure of β-MnO2 catalysts and their catalytic performance for ORR [212]. As shown in Fig. 14, a moderate concentration of bulk OVs will greatly increase the electric conductivity of MnO2, while excessive OVs will hinder the ORR process. Such a curvilinear relationship between the electronic structure and OV concentration suggests that the conductivity and ORR catalytic activity of β-MnO2 can be modulated by the OV concentration. Defect engineering can also be applied to increase the density of active sites of nanomaterials for electrocatalysis [203,213,214]. Xie et al. [203] designed a reaction with a high concentration of precursors and different amounts of thiourea, thus realizing controllable defect modulation in as-formed MoS2 ultrathin nanosheets. Due to the defect-rich structure, many tiny cracks formed on the basal surfaces, resulting in 13 times more active sites for the defect-rich MoS2 ultrathin nanosheets than for the defect-free MoS2.
《Fig. 14》
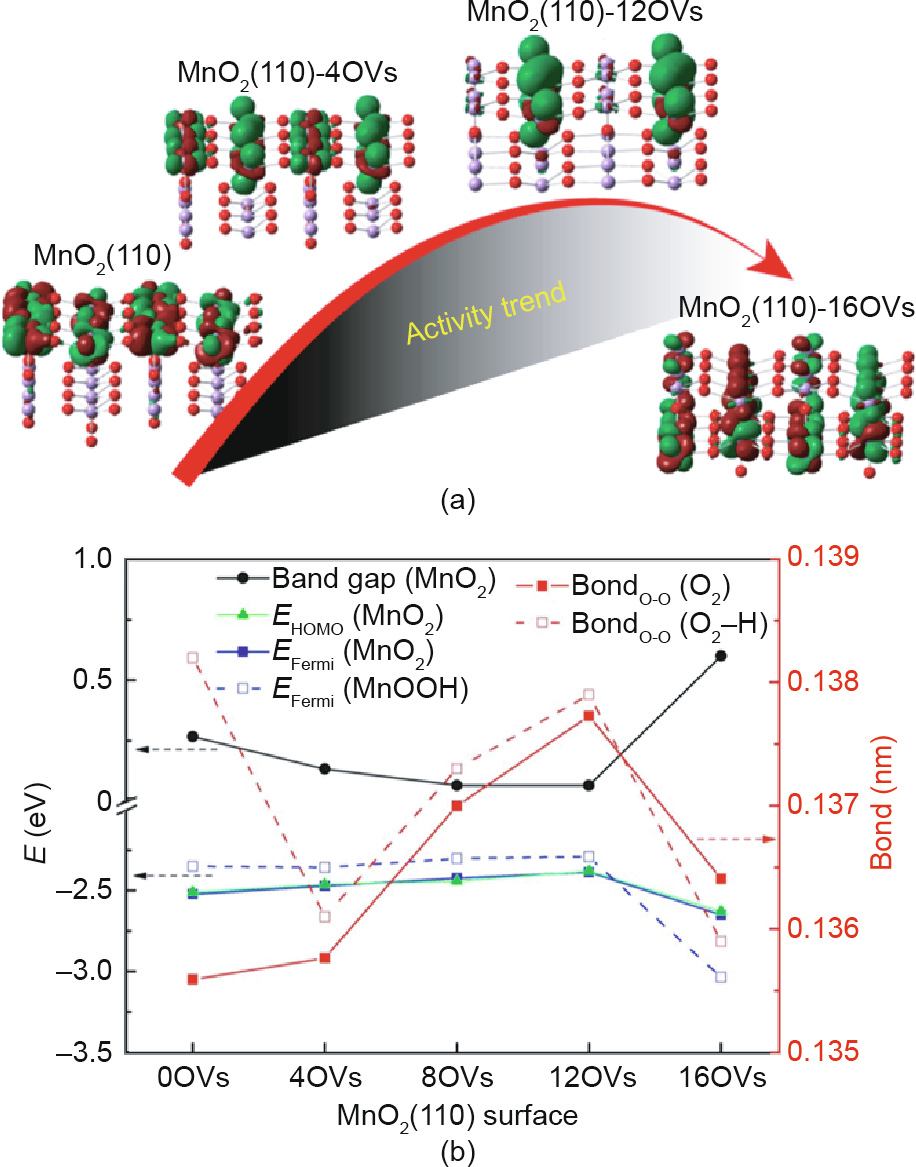
Fig. 14. (a) Activity trend of MnO2 with different OV concentrations; (b) changes of DFT results as a function of OV concentration. HOMO: highest occupied molecular orbital. Reproduced from Ref. [212] with permission of ACS Publications, ©2015.
Similar to element vacancy in metal compounds, intrinsic defects in carbon-based electrocatalysts are universal but have been ignored for a long time [209]. Defects easily form after heteroatom doping, and act as the active sites favoring electrocatalysis [215,216]. However, the electrocatalytic reactivity of carbon-based materials has mainly been ascribed to the induced changes of heteroatoms doping. As time goes on, some research has found that the catalytic activity of carbon electrocatalysts with intrinsic defects is even better than that of heteroatoms-doped carbon materials [217,218]. For example, Jiang et al. [219] found that defective carbon nanocages (CNC) possess a high ORR activity that exceeds that of B-doped carbon nanotubes. In this case, defect-rich CNC were successfully synthesized with many typical defect locations, but without any dopants (Fig. 15(a)). The electrochemical results indicated that the resultant CNC material with the highest defect density showed the best electrochemical ORR activity (Fig. 15(c)). The DFT results further indicated that the high ORR activities of these defect materials could be attributed to the pentagon and zigzag edge defects (Fig. 15(d)). Zhao et al. [220] used first principles calculations to predict that a type of 585 defect on graphene would be even more active than the N-doped sites for ORR, and obtained strong support for this theoretical prediction through experimental investigations. With the defect mechanism in mind, Zhao et al. [221] prepared a porous carbon (PC) material lacking any elemental doping by carbonizing Zn-MOF at 950 °C. With the benefit of the removal of zinc (Zn) atoms, defects could be formed on the PC catalyst, endowing the PC catalyst not only with excellent ORR activity, but also with a stability comparable to that of a commercial Pt/C catalyst. Furthermore, with the exception of the ORR process, the individual electrocatalytic activities for the other three electrochemical reactions in the energy conversion from water to water—that is, HOR, OER, and HER—were demonstrated to be particularly sensitive to the types of defects derived by the removal of heteroatoms from graphene [222].
《Fig. 15》

Fig. 15. (a) HRTEM (high resolution TEM) image and (b) schematic structure of CNC700; (c) rotating disk electrode (RDE) curves of these CNC samples; (d) ORR free-energy diagrams of different defects. Reproduced from Ref. [219] with permission of ACS Publications, ©2015.
《3.6. Atomic doping》
3.6. Atomic doping
Atomic doping is the most widely used strategy for modulating the properties of catalytic materials. By reasonably introducing one or more metallic or nonmetallic elements into the lattice of the material, the electron structure of the original material can be adjusted, thus effectively improving the catalytic performance of the material [223–232]. Taking MoS2 as an example, many metallic elements such as Ni, Co, Fe, vanadium (V), lithium (Li), and copper (Cu) have been reported to be successfully doped into its crystal structure, positively affecting the physical and chemical properties [171,233–237]. Among these doped metallic elements, Ni and Co tend to locate around the S in MoS2, which will decrease the hydrogen adsorption energy at the S edge and increase the density of the active sites in MoS2 [233–235]. Unlike Ni- and Co-doped MoS2, V doping cannot increase the number of active sites, but will enhance the conductivity of MoS2 [236]. Interestingly, our group explored the influence of the Ni-doping of molybdenum carbide on its surface electronic structure and its relationship with HER performance by combining experimental and theoretical evidence [97]. As shown in Figs. 16(a)–(d), one-dimensional (1D) NiMo2C nanowire arrays were directly constructed onto conductive 3D Ni foam (NiMo2C/NF) via a facile and controllable strategy combined with hydrothermal and post-carburization treatment. The binder-free integrated NiMo2C/NF electrode showed superb HER catalytic activity in comparison with Mo2C and Ni catalysts (Fig. 16(e)). The DFT calculations clearly demonstrated that the incorporation of Ni into the Mo2C lattice brought about changes in the charge distribution on the catalyst, which resulted in a synergistic effect of Ni and Mo2C that decreased the hydrogen binding energy (Figs. 16(f) and (g)).
Apart from metallic elements, research on doping with nonmetallic elements is also very active. Xie et al. [238] successfully synthesized oxygen-doped MoS2 ultrathin nanosheets, on which the synergistic modulations of both the active sites and the conductivity could be rationally realized. According to the DFT calculations, the smaller differential binding free energy of the oxygen-incorporated MoS2 revealed a lower energy barrier for driving the HER process. Our group further proposed a partial phosphorization of metal oxide precursors to construct oxygenincorporated NiMoP2 with enhanced HER activity [239]. As illustrated in Figs. 16(h)–(i), the H adsorption energy on the NiMoP2 surface was optimized by the oxygen incorporation, as the 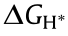 of the O–NiMoP2 is much closer to zero than its undoped equivalent. In addition, the Ni and Mo in O–NiMoP2 possessed more positive charge, which was beneficial for adsorbing and activating water molecules, greatly accelerating the water dissociation in the alkaline HER.
of the O–NiMoP2 is much closer to zero than its undoped equivalent. In addition, the Ni and Mo in O–NiMoP2 possessed more positive charge, which was beneficial for adsorbing and activating water molecules, greatly accelerating the water dissociation in the alkaline HER.
《Fig. 16》

Fig. 16. (a–d) SEM, TEM, and EDX (energy dispersive X-ray spectroscopy) images and (e) electrochemical characterization of NiMo2C electrodes; distribution of Bader charge of (f) Mo2C(001) and (g) NiMo2C(001); (h) hydrogen free-energy and (i) charge density distributions on different samples. (a–g) reproduced from Ref. [97] with permission of Royal Society of Chemistry, ©2015; (h, i) reproduced from Ref. [239] with permission of ACS Publications, ©2018.
Aside from these metal compounds, carbon-based materials have been intensively used as doping objects, greatly enlarging the scope of catalyst research [240–245]. Early in 2013, our group reported a phosphorus (P)-doped graphene with an ORR catalytic performance comparable to that of commercial Pt/C [246]. Furthermore, N, P dual-doped graphene/carbon materials were prepared as electrocatalysts for both ORR and OER, and their catalytic activities exceeded those of the benchmark Pt/C catalyst [247]. In order to reveal the underlying reasons for the high activity of the heteroatom-doped carbon, our group conducted a comprehensive DFT calculation on graphene doped by a series of different heteroatoms for ORR [248]. The DFT results indicated that there was a triple effect of the carbon sites—namely, the charge, spin density, and ligand effect—determining the intrinsic catalytic activity of the doped carbon catalysts and their ORR mechanism (Fig. 17). When the carbon materials are doped by a single heteroatom, the carbon sites around the doped atom can only be activated by the triple effect separately. This causes the ORR to proceed via the associative mechanism, and there is a limitation with an intrinsic overpotential of 0.44 V. However, when carbon materials are doped by metal or dual-heteroatoms, the ORR follows the dissociative mechanism, as double carbon sites can be activated by the triple effect. Thus, the activity limitation of the associative mechanism will no longer be in effect, leading to enhanced ORR activity. Our group also synthesized graphene co-doped with metallic and nonmetallic elements, and revealed the roles of nitrogen configuration in Ndoped graphene as well as that of the trace atomic Ni in HER [249]. We found that quaternary nitrogen (N) is the most active site of the three N types in HER, whereas when doping with trace atomic cobalt, the planar (pyridine and pyrrolic) N becomes the most active. In contrast, when trace atomic Co was replaced by Ni, the planar (pyridine and pyrrolic) N exhibited depressed HER activity.
《Fig. 17》
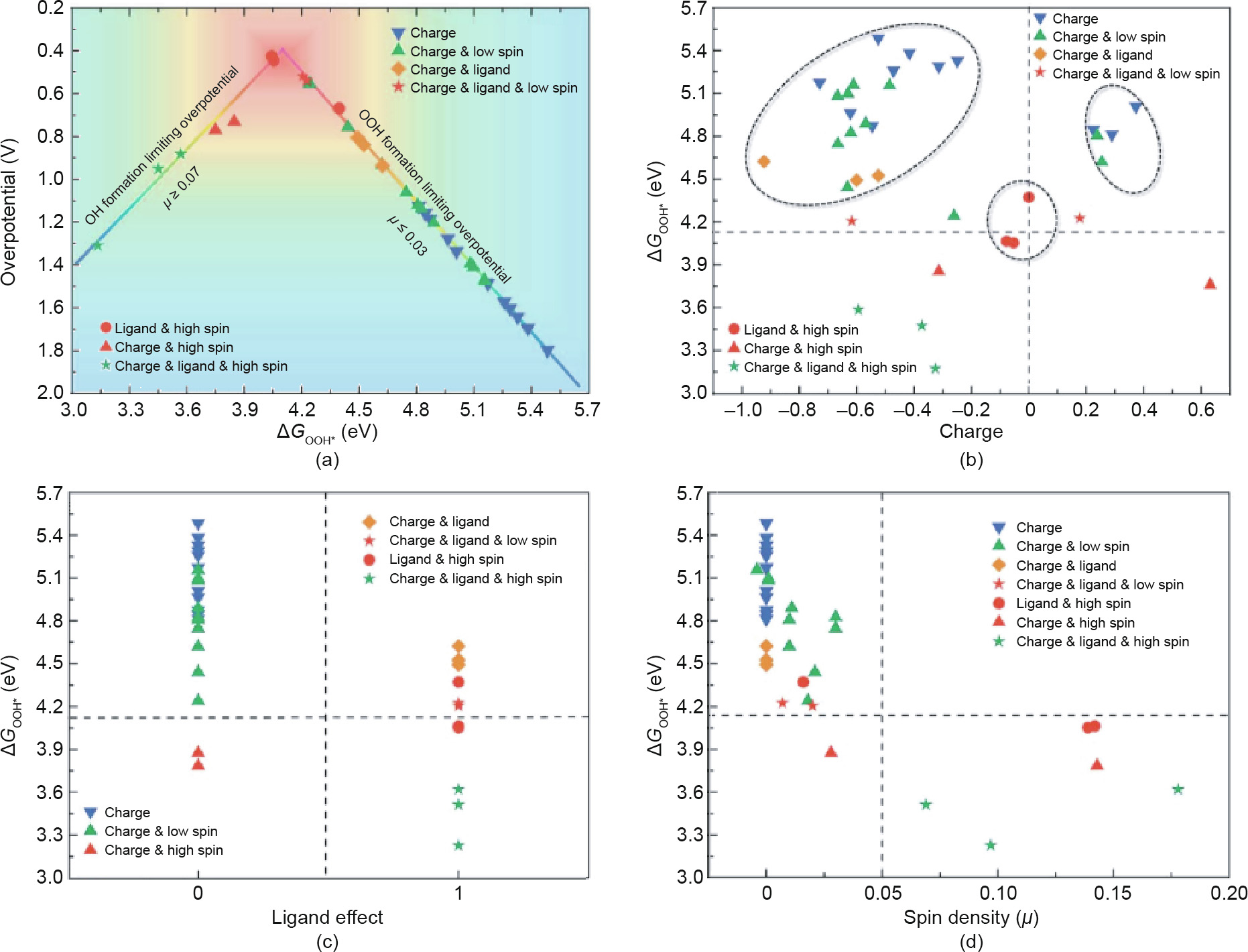
Fig. 17. (a) ORR overpotential as a function of  for carbon active sites. vs (b) charge effect, (c) ligand effect, and (d) spin density effect. Reproduced from Ref. [248] with permission of The Royal Society of Chemistry, ©2018.
for carbon active sites. vs (b) charge effect, (c) ligand effect, and (d) spin density effect. Reproduced from Ref. [248] with permission of The Royal Society of Chemistry, ©2018.
《3.7. Interface engineering》
3.7. Interface engineering
Hybrid nanomaterials have an interface located at the boundary of two components [250]. It is extremely important for a heterogeneous catalyst to have a proper interfacial structure because the interface region always presents unique physical and chemical properties [251]. These unique properties can facilitate the capability of the resultant material to bind, convert, and transport the surface species (e.g., adsorbents, electrons, and intermediates), greatly promoting the catalytic reactions occurring on the surface [252– 255]. In recent years, abundant research studies have reported the design and synthesis of electrocatalysts for water–hydrogen electroconversion with the assistance of interface engineering. In general, depending on the relative locations of the components, hybrid materials can be classified as supported structures, heterostructures, or core–shell structures [256]. The special character of a supported structure is that the support component is much larger than the other components; in contrast, the components in a heterostructured material have similar sizes. In a core–shell structure, one component is covered by another component, with interfaces existing at the boundary between the two components. These three types of hybrid materials with different interface structures have been reported as a result of research assembling metals, metal oxides, nonoxides, and so forth. For example, Zhang et al. [257] reported on a MoNi4 electrocatalyst immobilized on MoO2 cuboids (MoNi4/MoO2@Ni) with a supported structure that was made by controlling the outward diffusion of nickel atoms during calcination. By heating the NiMoO4 precursor under a reduction atmosphere, MoNi4 nanoparticles (20–100 nm) supported by MoO2 cuboids (~1 μm) were synthesized, and the supported hybrid catalyst was found to exhibit excellent HER activity in alkaline solution (Figs. 18(a) and (b)). Heterostructured Co/CoP nanoparticles were prepared by Xue et al. [258] via the gradual phosphidation of Co metal into CoP components. By changing the weight ratios of the NaH2PO2 and Co species, the CoP contents in Janus Co/CoP nanoparticles could be controllably modulated, affecting the interface zone in the Co/CoP catalyst (Figs. 18(c)–(f)). As illustrated in Figs. 18(g) and (h), Xu et al. [259] fabricated NiCo-based porous microrod arrays composed of carbon-confined NiCo@NiCoO2 core@shell nanoparticles (NiCo@NiCoO2/C PMRAs (porous microrod arrays)) by the reductive carbonization of bimetallic (Ni, Co) metal–organic framework microrod arrays and subsequent controlled oxidative calcination. The obtained NiCo@NiCoO2/C PMRAs combined several desirable qualities for OER, including a large surface area, good conductivity, and multiple electrocatalytic active sites.
《Fig. 18》
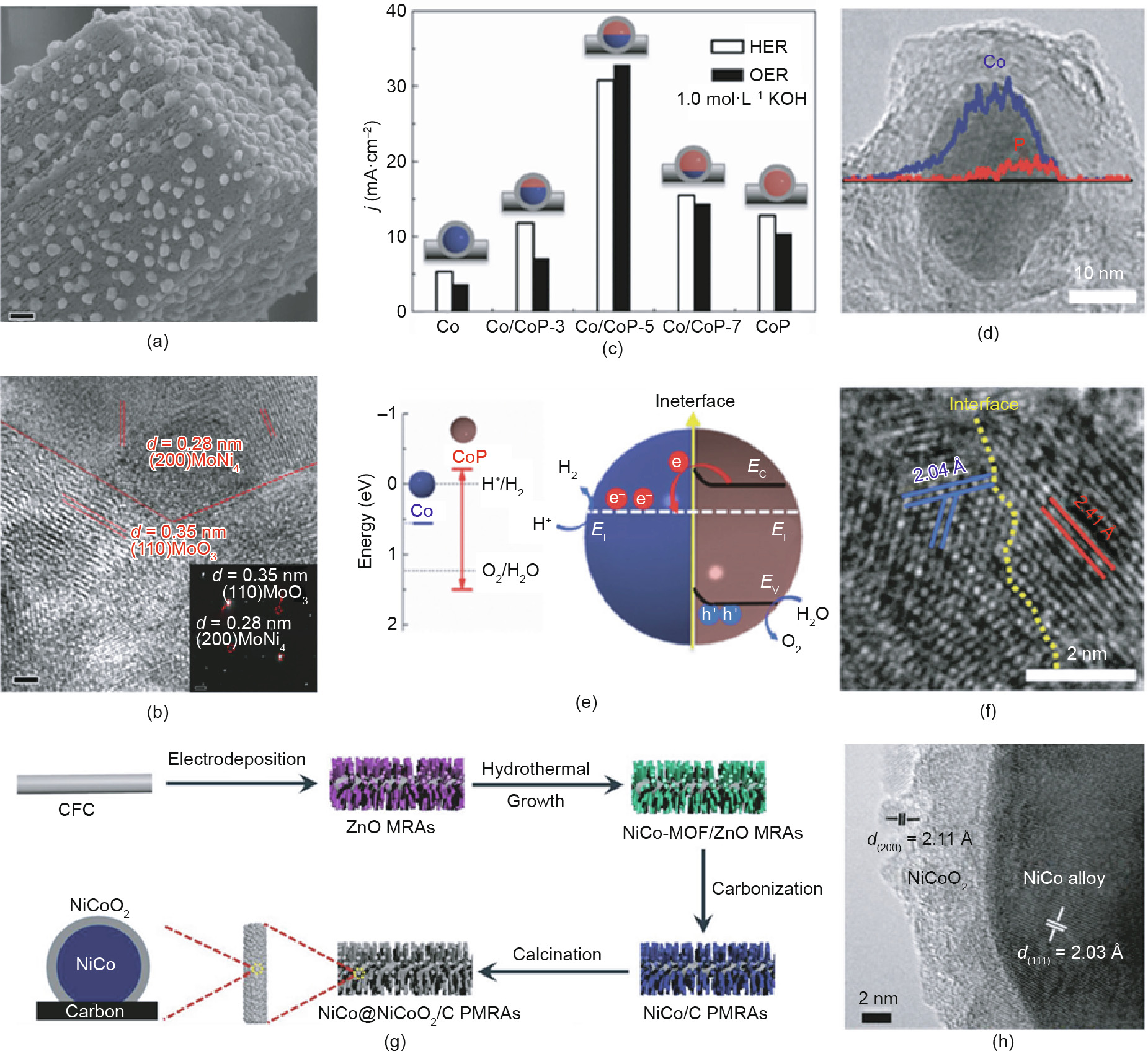
Fig. 18. (a, b) Typical SEM images of MoNi4/MoO2@Ni; (c) current densities of the obtained samples; (d) compositional line profile of a typical Co/CoP-5 nanoparticle; (e) electronic structures of metallic Co and CoP, and the Co/CoP-based Mott-Schottky contact; (f) HRTEM image of a typical Co/CoP-5 nanoparticle; (g) fabrication process and (h) HRTEM image of NiCo@NiCoO2/C PMRAs. CFE: carbon fiber cloth; MRA: microrod arrays. Reproduced from Ref. [259] with permission of WILEY-VCH Verlag GmbH & Co. KGaA, ©2018.
For the purpose of rational catalyst design, the origin of the enhanced catalytic performance of these hybrid materials with an abundant interface has been deeply explored. The modulation of the electronic structure in the interface of hybrid materials has been well proven [260–267]. An observed electron transfer was evidenced by Yu et al. [268] using X-ray absorption near-edge spectroscopy (XANES) for a Ni(OH)2/Pt catalyst. As pointed out by the authors, the pre-edge and main absorption edge of Ni for both the α- and β-Ni(OH)2/Pt electrode are shifted to lower energies, and the absorption intensities decrease in comparison with their counterparts, demonstrating that the electrons transfer from the Pt substrate to the hydroxide. Han et al. [269] revealed a strong metal-support interaction (SMSI) effect between CoS2 and Pt in the Pt/CoS2 hybrid system for highly active water splitting. A downward shift in the Pt d-band structure was proved by DFT calculation and was further supported by X-ray photoelectron spectroscopy (XPS) and X-ray absorption fine structure (XAFS) analyses. Although the electron transfer has been well demonstrated in these metal/metal compound catalysts, the underlying mechanisms for the high catalytic activity are ambiguous. In order to uncover this mystery, our group studied the chemical properties of the metal/metal oxide interface, and found an interface-induced synergistic effect—the ‘‘chimney effect” (Figs. 19(a) and (b)) [270]. As revealed by the DFT results (Figs. 19(c) and (d)), the neighboring sites around the interface are immune to H2O* and OH* species but selectively adsorb H*, which prevents the poison effect of H2O* and OH* on active sites. Meanwhile, the active sites on the interface have good capability for the adsorption and desorption of the H* reactant for the H2 product, as its 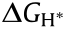 is close to 0. Due to these features, the hydrogen evolution on metal oxide/metal catalysts behaves similarly to a chimney for generating hydrogen productive continuously. The strong positive correlation between the HER activity of the metal oxide/metal composites and the interfacial metal atoms (Fig. 19(e)) further confirmed that the hydrogen generation can be greatly accelerated by increasing the amount of active site. With the exception of metal/metal compound catalysts, the origin of the performance enhancement of metal compound hybrid catalysts was further investigated by our group through the casting of monometallic NiO–Ni3S2 heteronanosheets [271]. The electron transfer from Ni–S to Ni–O that occurs at the interface of NiO–Ni3S2 results in a superior activity of overall water splitting that is even superior to the benchmark Pt/C and RuO2 assembly (Fig. 19(g)). DFT calculation results further demonstrated that the activation barrier of the hydrogen (or oxygen-containing) intermediates on the NiO–Ni3S2 interfaces are significantly lowered, which is beneficial for the OER and HER processes (Figs. 19 (h) and (i)).
is close to 0. Due to these features, the hydrogen evolution on metal oxide/metal catalysts behaves similarly to a chimney for generating hydrogen productive continuously. The strong positive correlation between the HER activity of the metal oxide/metal composites and the interfacial metal atoms (Fig. 19(e)) further confirmed that the hydrogen generation can be greatly accelerated by increasing the amount of active site. With the exception of metal/metal compound catalysts, the origin of the performance enhancement of metal compound hybrid catalysts was further investigated by our group through the casting of monometallic NiO–Ni3S2 heteronanosheets [271]. The electron transfer from Ni–S to Ni–O that occurs at the interface of NiO–Ni3S2 results in a superior activity of overall water splitting that is even superior to the benchmark Pt/C and RuO2 assembly (Fig. 19(g)). DFT calculation results further demonstrated that the activation barrier of the hydrogen (or oxygen-containing) intermediates on the NiO–Ni3S2 interfaces are significantly lowered, which is beneficial for the OER and HER processes (Figs. 19 (h) and (i)).
《Fig. 19》

Fig. 19. (a) Schematic diagrams of two possible HER pathways on RuO2/Ni composite catalysts; (b) a “chimney effect” at the metal/metal oxide interface; (c) the curve of vs HBE (hydrogen binding energy); (d) adsorption energy of H2O*, OH*, and H* species on different clusters; (e) variation trend of the Niinterface content and the HER activities of NiO/Ni samples; (f) schematic of water electrolysis catalyzed by monometallic NiO–Ni3S2 heteronanosheets; (g) polarization curves of the NiO–Ni3S2 couple or Pt/C and RuO2 electrode couple for water electrolysis; (h) hydrogen free energy and (i) OER free-energy diagrams over Ni3S2 and O–Ni3S2 surfaces. (a–e) reproduced from Ref. [270] with permission of Elsevier B.V., ©2019; (f–i) reproduced from Ref. [271] with permission of Elsevier Inc., ©2019.
Vacancy generation is another important factor in developing interfaced materials with improved catalytic performances [272– 277]. A typical example was reported by Li et al. [278], who prepared FeS2/CoS2 hybrid nanosheets (NSs) for overall water electrolysis. In the synthesis process, CoFe2O4 nanoparticles were first synthesized by a coprecipitation method, and then transformed into the FeS2 and CoS2 phases. During the latter sulfuration process, interfaces with defect sites were created in the FeS2/CoS2 NSs. Electron paramagnetic resonance (EPR) spectra revealed a stronger EPR signal at g = 2.007 recorded with the FeS2/CoS2 NSs composite, indicating abundant S vacancies. Extended X-ray absorption fine structure (EXAFS) was used to further investigate the local structure of the obtained samples, and a distinct decrease in peak intensity in Fe Kedge EXAFS was detected with FeS2/CoS2 NSs, demonstrating the coordination deficiency from Fe. Similar results were reported by Gao et al. [279] in CeO2/NiO with an embedded configuration (Ce–NiO–E) or the surface-loaded configuration (Ce–NiO–L). An increasing trend of Ni3+ and oxygen defects for NiO (Ni3+62%, O defect 24%), Ce–NiO–L (Ni3+ 69%, O defect 26%), and Ce–NiO–E (Ni3+ 71%, O defect 32%) correlated well with the activity trend, indicating that these vacancy sites at the interface region contribute greatly to the enhanced activity. In fact, electron modulation and vacancy do not exist independently, and the catalytic performances of the interfaced materials may be influenced by the two factors simultaneously.
《3.8. Alloying》
3.8. Alloying
Alloying is the diffusion penetration of the metal atoms of two or more metals, or the addition of nonmetallic elements into metals through melting, sintering, or vapor deposition processes. Metal alloying is an effective strategy for improving the performance of metal catalysts. It can not only refine the grain size to improve the mechanical strength and surface area of catalysts, but also selectively reduce the amount of a single component (e.g., Pt, Au, Ru, and other precious metals) in order to reduce the catalyst cost. Moreover, the catalytic activity and selectivity of metals can be changed by incorporating other metals to form alloys, due to the synergistic effect between metal components [280–284]
According to the Brewer–Engel valence bond theory [285], alloying a metal with an unfilled d orbital and a metal with internally paired d-electrons can modulate the hydrogen adsorption energy on the alloy surface, thus improving the hydrogen evolution activity. Raj [286] prepared a series of Ni-based binary composites by means of electrodeposition techniques. The trend in their HER activities in alkaline water followed this order: Ni–Mo > Ni–Zn >Ni–Co > Ni–W > Ni–Fe > Ni–Cr. Due to its excellent activity, the Ni–Mo alloy has been considered to be the most promising HER catalytic material. Zhang et al. [287] successfully constructed a layer of Ni–Mo alloy nanorods with uniform size and uniform element distribution on the surface of a nickel substrate by means of magnetron sputtering technology, and found that the HER exchange current density was almost ten times higher than that of single-metal Ni or Mo catalysts. This research showed that the excellent catalytic activity of the Ni–Mo alloy electrode mainly comes from two factors: ① There is an increase in the specific surface, caused by the grain refinement of the two-component metal in the growth process; and ② the electronegativity difference between the elements Ni and Mo causes electrons to concentrate around Mo, thus forming a synergy for electrocatalysis.
In order to reduce the usage of precious metals, many nonnoble metals (i.e., Co, Ni, Fe, Cu, V, Cr, Mn, Zn, etc.) have been incorporated with noble metals to form alloys as electrocatalysts [206,288]. Studies of ORR performances on PtM alloys revealed the following activity order [289–291]: PtFe/C > PtCo/C > PtV/C > PtNi/C > Pt/C. The following stability order was also determined [292]: Pt3Ir(111) > Pt3Co(111) > Pt3Ni(111) > Pt3Fe(111). In addition, Greeley et al. [293] found a classic volcano-shaped dependency based on the ORR activity of Pt alloys versus the d-band center position of 3d metals (Fig. 20). According to that study, the ORR mechanism on Pt3M catalysts was either O2 dissociation or proton/electron transfer to molecular O2, and the optimal ORR catalyst should have a weaker oxygen-binding energy than Pt. Bampos et al. [294] further synthesized a series of carbonsupported Pd–M (where M = Ag, Co, Cu, Fe, Ni, or Zn) bimetallic catalysts as ORR electrocatalysts in acidic solution. The ORR activity of these Pd-based alloys was found to descend as follows: PdZn/C > PdNi/C > Pt/C > PdAg/C ≥ PdCo/C > PdFe/C > PdCu/C > Pd/C. Notably, PdZn/C exhibited a specific activity three times higher than Pt/C at a potential between 0.35 and 0.5 V versus Ag/AgCl.
《Fig. 20》
Fig. 20. (a) Relationship between the measured kinetic current density and the calculated oxygen adsorption energy,  ; (b) of Pt on the surface of Pt-based alloys vs alloying energy [293]. Reproduced from Ref. [26] with permission of Springer Nature Limited, ©2009.
; (b) of Pt on the surface of Pt-based alloys vs alloying energy [293]. Reproduced from Ref. [26] with permission of Springer Nature Limited, ©2009.
In addition to metallic elements, the introduction of nonmetallic elements can improve the catalytic performance of alloys [295– 299]. For example, Kiran et al. [296] prepared few-layer MoS2(1–x )Se2x alloys that possessed higher HER activity than pristine MoS2 and MoSe2. A systematic structure-activity relationship was revealed by tuning the Se/S incorporation in MoS2(1–x )Se2x , with MoS1.0Se1.0 having an Se/S ratio of 1:1 showing the highest HER activity. Similar results regarding Mo–S–Se alloys were reported by the groups of Gong et al. [299] and Xu et al. [297]. Xu et al. [298] successfully controlled the composition of sulfur (S) and selenium (Se) in ternary WS2(1–x )Se2x nanotubes on carbon fibers. The disordered atom arrangement introduced in the alloyed structure resulted in WS2(1–x)Se2x having excellent electrocatalytic properties for HER. A ternary pyrite-type CoPS was further obtained by Cabán-Acevedo et al. [300] for photo/electrochemical hydrogen evolution. Because of the higher electron-donating character of the P2– ligands in CoPS, this ternary pyrite CoPS was endowed with a more thermoneutral hydrogen adsorption, resulting in a higher HER activity than CoS2.
《4. Conclusions and perspectives》
4. Conclusions and perspectives
The majority of the energy dissipation in electrochemical hydrogen–water conversion is induced by the activation energy driving the electrochemical reactions involved in the energy system. Materials are at the core of a high-efficiency energy system, as the activation energy of the reactions involved in the system is strongly influenced by the catalytic materials. It is urgent to accelerate the development of active and robust electrocatalysts with acceptable cost in order to ensure that an efficient and sustainable energy system becomes a reality. In general, a catalyst’s performance depends on two main factors: ① the number of active sites in a given area; and ② the intrinsic activity of each active site. Tuning the geometric structure (i.e., the nanoarchitecture and facet engineering) makes it possible to realize enhancement of the active sites, because when the catalyst size is reduced to the nanoscale, the catalyst possesses a high surface area with increased exposure of the catalytically active planes. Defect engineering and amorphization can also be applied to enrich active sites by exposing thermodynamically unstable active sites or active unsaturated atoms. Increasing the intrinsic activity of the active site is a more fundamental yet difficult strategy for activity improvement, which involves accurate modulation of the electronic structures of catalytic materials. Optimization of the intrinsic activity can be achieved based on a fundamental understanding of the particularities of the reactions and insight into the design of catalysts with targeted functionalities. The featured examples (their catalytic performances are summarized in Tables 2–5) discussed herein present successful modulation of the intrinsic activity via element doping, interface engineering, polymorph engineering, alloying, and so forth, with the aid of synergistic interaction between theoretical and experimental studies.
《Table 2》
Table 2 Summary of electrocatalysts’ performance in HER.

CNT: carbon nanotube; DG: defect graphene; HDs: Heterodimers; NPLs: nanoplates; PBS: phosphate buffer saline; TM: Ti mesh; MMT: montmorillonite; SR: -SCH2CH2Ph.
《Table 3》
Table 3 Summary of electrocatalysts’ performance in OER.

GCNS: graphene/carbon nanosheets; MMO: mixed metal oxide.
《Table 4》
Table 4 Summary of non-noble metal electrocatalysts’ performance in ORR.
CF: carbon nanofibrous; MCSs: mesoporous carbon microspheres; NCNT: nitrogen-doped carbon; Nanotube; OMC: Ordered mesoporous carbons; OMMC: ordered macro-/ mesoporous carbon; PoPD: porous N-doped carbon.
《Table 5》
Table 5 Summary of noble-metal electrocatalysts’ performance in ORR.

OFA: octopod nanoframe architectures; TOH: trisoctahedrons.
At present, with the rapid development of chemical synthesis techniques, many catalytic materials have been reported with outstanding electrocatalytic performance. Meanwhile, developments in physical characterization methods and theoretical calculation are providing more evidence and guidance for researchers, allowing us to better understand the performance-improvement mechanism of catalysts. However, the new-generation electrode catalysts are mainly being produced through traditional trialand-error processes rather than by means of rational design. This research model not only wastes social resources and the energy of scientific researchers, but also greatly delays scientific development. Thus, the development of a performance-oriented design strategy for electrocatalyst design from the most fundamental level to a practical application level is urgent. This target requires computational chemistry, electrocatalytic chemistry, and synthetic chemistry working in concert; unfortunately, however, all three fields require improvement at present. Current theoretical calculation models are still imperfect, as most do not take into account the influence of ionic strength, the double-layer effect, the solvation effect, and so forth, making it difficult to accurately reflect the actual reaction process of the catalytic material surface. Furthermore, although the reaction mechanisms of these hydrogen- and oxygen-involving reactions have been widely investigated, the actual mechanisms on different catalyst surfaces remain shrouded in mystery. In fact, it is also difficult to identify the actual active sites, as the surface structure of most catalysts varies during the electrocatalytic process. New progress will come from the merging of advanced theoretical calculations and experimental characterization (e.g., in situ, ex situ, and operando techniques), which will allow us to promote our understanding of the electrochemical reaction mechanism and the dynamic evolution of the catalysts involved at the molecular level. Finally, function-oriented catalyst preparation remains a major challenge. Existing material synthesis technologies are able to regulate the physical and chemical properties of specific catalysts to some extent at the nanoscale, but the use of controlled synthesis technology to modulate the electronic structure of catalysts at the atomic scale remains immature. The development of controllable synthesis methods with strong applicability and large-scale production is another focus of future research.
《Acknowledgements》
Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (21576032 and 51772037), the Key Program of the National Natural Science Foundation of China (21436003), the Major Research Plan of the National Natural Science Foundation of China (91534205), and the National Program on Key Basic Research Project of China (2016YFB0101202).
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Lishan Peng and Zidong Wei declare that they have no conflict of interest or financial conflicts to disclose.
《Nomenclature》
Nomenclature
c reactant surface concentration, mol·m-3
F Faraday constant, ~96 485 C·mol-1
G Gibbs free energy, J·mol-1
f decay rate to products and reactants, dimensionless
i current, A
j current density, A·cm-2
j0 exchange current density, A·cm-2
n number of electrons transferred in the reaction, dimensionless
R resistance, Ω
T temperature, K
U0 reaction equilibrium potential, V
V voltage, V
α charge transfer coefficient, dimensionless
μ overvoltage, V
 activation energy barrier, J·mol-1
activation energy barrier, J·mol-1
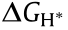 hydrogen adsorption free energy
hydrogen adsorption free energy
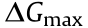 gibbs free-energy change, J·mol-1
gibbs free-energy change, J·mol-1
 oxygen adsorption strength
oxygen adsorption strength











 京公网安备 11010502051620号
京公网安备 11010502051620号




