

《1. Introduction》
1. Introduction
Since Friedrich Wöhler’s pioneering synthesis of urea in 1828, increasingly sophisticated techniques for total synthesis of organic compounds [1] have been developed and advanced to meet the challenges posed by ever more diverse and complex molecular targets. Total synthesis has made substantial contributions to the replication, confirmation, disproving, and revision of natural products and to the design and synthesis of drugs [2], benefiting not only the science community but also society. For example, the discovery of novel lead compounds is central to the process of drug design. Ligand-based and structure-based virtual screening techniques are the most common procedures used to identify the lead compounds that are most likely to bind to a protein target, via a rapid in silico evaluation of compounds in existing libraries. However, virtual screening techniques suffer from two drawbacks. First, the compounds identified already exist, leaving little room for new patents. Second, it is increasingly challenging to retrieve a new compound from existing libraries, considering that the chemical structural space explored in existing libraries is very limited in comparison with the theoretical space of all possible compounds. These limitations have led to a proliferation of studies in de novo drug design, which directly generates novel molecular structures to match desired pharmacological properties. While de novo design brings the benefits of easily patenting generated compounds and exploring the whole chemical space, which comprises on the order of 1060–10100 compounds [3], synthesizing de novo designed molecules, which are usually non-existing, is the main obstacle. In fact, synthetic accessibility (i.e., the ease of synthesis of compounds) can be a critical issue for the compounds identified by virtual screening; that is, an identified compound that is not found in off-the-shelf catalogs and not purchasable requires synthesis.
Retrosynthesis prediction, which was first formalized by Corey and Wipke [4] in 1969, set in motion a revolution of total synthesis with increased rationality and systematicness. It designs a sequence of reactions to recursively decompose a target compound into simpler building blocks until commercially available starting molecules are reached. The retrosynthesis prediction framework consists of two basic tasks: single-step retrosynthesis prediction and multi-step retrosynthesis prediction. The primary goal in single-step retrosynthesis prediction is to predict possible reactants given a target molecule, as shown in Fig. 1. By means of disconnection, which breaks the bonds between the atoms of the target molecule, one or more synthons are obtained. It should be noted that a synthon (e.g., the electrophilic ‘‘PhC=O+ ” group in Fig. 1) as a fragment is an ion or radical and usually does not exist as a stable species, whereas the reactant (e.g., benzoyl bromide) corresponding to the synthon is an actual chemical used in practice for synthesis. Once accurate and recursive single-step retrosynthesis prediction is complete, multi-step retrosynthesis prediction focuses on planning the optimal reaction sequence that minimizes the number of synthesis steps, the cost of the starting molecules, the waste produced, and so forth.
《Fig. 1》

Fig. 1. An example illustration of a single-step retrosynthesis reaction. By breaking one bond in the given product, two synthons come into being, where the left one carries a positive charge and the right one carries a negative charge. After rearrangement toward real substances, the two reactants are obtained.
The retrosynthesis prediction problem is particularly challenging. First, the number of possible chemical transformations is huge, resulting in a vast search space. Consequently, extensive creativity and expertise from experienced chemists are required. Second, even for human experts, current understanding of reaction mechanisms remains incomplete, and it is non-trivial to select the globally optimal route among numerous possible retrosynthesis routes. Since the 1960s, a great deal of focus and expectation have been placed on computer algorithms to assist experienced chemists in retrosynthesis designs. Rule-based expert systems [5,6] have been the most widely investigated approach for many years. The core of rule-based expert systems lies in the use of retrosynthesis templates. Each template, as shown in Fig. 2, is represented by molecular subgraph patterns that encode changes in the connectivity of atoms during a chemical reaction. Starting from a target molecule, a template is selected following predefined rules and is applied to the target molecule to determine the reactants.
《Fig. 2》

Fig. 2. Illustration of a chemical reaction and its retrosynthesis template. The highlighted parts represent the reaction centers of the chemical reaction; molecular subgraph patterns of the reaction centers are used to compose the corresponding template. The atoms marked with stars are attached atoms outside the template in molecules.
Unfortunately, rule-based expert systems suffer from several drawbacks. The major criticism of such systems is their incapability to provide accurate predictions for novel target molecules or reaction types out of their knowledge base [7,8]. Another limitation is their limited scalability: Applying hundreds of thousands of templates, which can be seen as repeatedly solving the subgraph isomorphism problem [9], is computationally expensive, as has been pointed out in many works [10,11]. Moreover, manually encoding the rules is laborious [6,12], although these programs can identify complex routes if given sufficient time investment [13].
Recently, the wide success of artificial intelligence (AI) has triggered a surge of interest in using machine learning techniques to overcome the limitations of rule-based expert systems. Here, the key principle is to have machines learn chemistry from large experimental datasets in order to predict reactions and perform retrosynthesis. To date, an extensive series of studies have demonstrated that AI-assisted models [13–15] can achieve competitive and even superior retrosynthesis planning performances in comparison with rule-based systems [5,6]. In the remaining part of this review, we discuss the existing AI algorithms for single-step and multi-step retrosynthesis prediction, respectively.
《2. The retrosynthesis problem》
2. The retrosynthesis problem
《2.1. Rigorous formulation of the problem》
2.1. Rigorous formulation of the problem
Before describing the literature, we first introduce the formal definitions and notations involved in the problem of retrosynthesis prediction:
(1) Molecule. A molecule with n atoms is characterized as a graph G = ( A, X ) , where 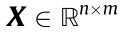 denotes the node features encoding the node type (e.g., H atom and
denotes the node features encoding the node type (e.g., H atom and  is the adjacency matrix. m and
is the adjacency matrix. m and  represent the number of node features and bond types, respectively.
represent the number of node features and bond types, respectively.  indicates that the ath node and the bth node have a bond of type c). It should be noted that the representation of a molecule can also be a simplified molecular input line entry system (SMILES) string [16] transformed from the graph.
indicates that the ath node and the bth node have a bond of type c). It should be noted that the representation of a molecule can also be a simplified molecular input line entry system (SMILES) string [16] transformed from the graph.
(2) Chemical reaction. A chemical reaction in this context is a transformation from a set of molecules to another set, that is,  where Ri denotes the ith reactant molecule and Pj denotes the jth product molecule; nr and np are the numbers of reactants and products, respectively.
where Ri denotes the ith reactant molecule and Pj denotes the jth product molecule; nr and np are the numbers of reactants and products, respectively.
(3) Single-step retrosynthesis prediction. The single-step retrosynthesis prediction problem is the reverse of a chemical reaction, as it predicts a set of reactants  that could produce a desired product P; that is,
that could produce a desired product P; that is,  . It should be noted that only single-outcome reactions with a single product P are considered here, as multi-outcome reactions can be decomposed into multiple single-outcome ones [10,17].
. It should be noted that only single-outcome reactions with a single product P are considered here, as multi-outcome reactions can be decomposed into multiple single-outcome ones [10,17].
(4) Reaction center. The reaction center includes the atoms and bonds that are directly involved in the bond and electron rearrangement of a reaction [18]. A reaction center is of the utmost importance to a chemical reaction, as given a specific reaction center, reactant prediction is much easier.
(5) Synthon. Disconnecting the bonds in the reaction center of the product P leads to a set of subgraphs  as synthons. A synthon Si is a subgraph of a reactant Ri and is not necessarily a valid molecule.
as synthons. A synthon Si is a subgraph of a reactant Ri and is not necessarily a valid molecule.
(6) Template. A retrosynthesis template T is denoted as the following rule [10]:

where pT is a subgraph of the product P and can be regarded as the reaction center, while  is the subgraph of the ith reactant.
is the subgraph of the ith reactant.
(7) Multi-step retrosynthesis planning. Provided with a target molecule P, multi-step retrosynthesis planning aims to predict a sequence of reactions  , where dmax is the length of the retrosynthesis pathway and rd is a single-step reaction, until all reactants required for P belong to a set of commercially available reactants
, where dmax is the length of the retrosynthesis pathway and rd is a single-step reaction, until all reactants required for P belong to a set of commercially available reactants  . If a sequence reaches the maximum number of retrosynthesis steps dmax but there remains an unsolved reactant that does not belong to the set , the planning is a failure.
. If a sequence reaches the maximum number of retrosynthesis steps dmax but there remains an unsolved reactant that does not belong to the set , the planning is a failure.
《2.2. Research challenges》
2.2. Research challenges
Fig. 3 demonstrates three exemplar retrosynthesis routes, the first two of which are successful, while the last one is a failure. To guarantee the accuracy and quality of retrosynthesis routes, we highlight the key research challenges in retrosynthesis planning below.
《Fig. 3》

Fig. 3. Demonstration of two successful routes and one failed route using retrosynthesis planning. It should be noted that all routes start from the starting materials and end with the target molecule. Chemists evaluate a route as successful if every reaction involved is chemically correct; otherwise, the route is a failure. In this case, the route fails because the reaction in the red rectangle is impossible. A, B, and C refer to the molecules synthetised in routes.
2.2.1. Single-step retrosynthesis prediction
Although only one-step reactions are generated in single-step retrosynthesis prediction, several challenges still exist. According to the definition of single-step retrosynthesis prediction provided in Section 2.1, machines encounter two main issues: ① how to determine the reaction center of the product P; and ② how to generate the reactants and reagents  after determining the reaction center. The issue of ‘‘how to determine the reaction center” involves determining what principle can be used to mimic a chemist’s brain when determining the disconnections for a target molecule. Determining the disconnections is challenging even for experienced chemists, as there are usually multiple ways to decompose a molecule, and the globally optimal synthetic route depends on global structures of the route [19]. Experts follow some basic rules [20] to intuitively determine the priority of chemical bond breaking. Unfortunately, these rules are particularly tedious, poorly generalized to various products, and even ill-defined due to the limit of existing chemistry knowledge. For machines, the oneto-many relationship in the identification of the reaction center (i.e., a target molecule P can have multiple reaction centers and be synthesized with many potential reactions) poses significant model-fitting and evaluation challenges.
after determining the reaction center. The issue of ‘‘how to determine the reaction center” involves determining what principle can be used to mimic a chemist’s brain when determining the disconnections for a target molecule. Determining the disconnections is challenging even for experienced chemists, as there are usually multiple ways to decompose a molecule, and the globally optimal synthetic route depends on global structures of the route [19]. Experts follow some basic rules [20] to intuitively determine the priority of chemical bond breaking. Unfortunately, these rules are particularly tedious, poorly generalized to various products, and even ill-defined due to the limit of existing chemistry knowledge. For machines, the oneto-many relationship in the identification of the reaction center (i.e., a target molecule P can have multiple reaction centers and be synthesized with many potential reactions) poses significant model-fitting and evaluation challenges.
Similarly, the other issue of ‘‘how to generate the reactants and reagents” involves determining which reactants and reagents are necessary for the reaction. Once the reaction center has been specified, a target molecule P can be broken into synthons  ; however, the synthons are not necessarily valid. Directly generating or converting these synthons into valid reactants and reagents
; however, the synthons are not necessarily valid. Directly generating or converting these synthons into valid reactants and reagents  is a formidable task that must meet three levels of validity. Firstly, the generated reactants must follow correct chemical grammar— that is, they must be ‘‘valid” molecules. Fig. 4(a) shows an ‘‘invalid” molecule. Secondly, the reaction from the reactants to the product must be chemically feasible; that is, the selectivity of the reaction center must be in accordance with reagents, reaction condition, electronic effects, steric hindrance, molecular orbital theory, and so forth. In Fig. 4(b), we show an infeasible reaction whose reaction center has low reactivity due to the electronic effects. Lastly, all atoms in the target product must be mapped to those in the reactants—that is, the reaction must follow the law of conservation of atoms. The reaction shown in Fig. 4(c) is a counterexample in which the law of conservation of atoms is not satisfied. More importantly, the method used for reactant generation varies by the representations of a molecule; graph-based methods are a top priority for graph representations, while sequence-based methods are a perfect match for SMILES representations.
is a formidable task that must meet three levels of validity. Firstly, the generated reactants must follow correct chemical grammar— that is, they must be ‘‘valid” molecules. Fig. 4(a) shows an ‘‘invalid” molecule. Secondly, the reaction from the reactants to the product must be chemically feasible; that is, the selectivity of the reaction center must be in accordance with reagents, reaction condition, electronic effects, steric hindrance, molecular orbital theory, and so forth. In Fig. 4(b), we show an infeasible reaction whose reaction center has low reactivity due to the electronic effects. Lastly, all atoms in the target product must be mapped to those in the reactants—that is, the reaction must follow the law of conservation of atoms. The reaction shown in Fig. 4(c) is a counterexample in which the law of conservation of atoms is not satisfied. More importantly, the method used for reactant generation varies by the representations of a molecule; graph-based methods are a top priority for graph representations, while sequence-based methods are a perfect match for SMILES representations.
《Fig. 4》

Fig. 4. Three types of error in a single-step retrosynthesis prediction. (a) An incorrect molecule in which a fluorine atom has three bonds; (b) an incorrect reaction center in which a bromine atom is incorrectly attached to the ortho position of the ester group; (c) a reaction that does not follow the conservation of atoms, as an additional carbon atom is added to the product but is not present in the reactant.
2.2.2. Multi-step retrosynthesis prediction
As the length of a retrosynthesis pathway can be as long as 60 or more steps [21], the ultimate goal for retrosynthesis planning is to generate a multi-step retrosynthesis route, which can be achieved by using the aforementioned one-step retrosynthesis prediction algorithms as a foundation. While one-step retrosynthesis predictions that reflect reaction feasibility have been widely investigated and continuously improved, fewer novel algorithmic attempts have been made to address the extremely challenging multi-step retrosynthesis prediction problem of navigating toward commercially available building-block materials, . Several challenges must be addressed when designing and evaluating a satisfactory retrosynthesis planning model. First, the search space for possible retrosynthesis plans over all possible synthesis routes is often exponentially large, considering that there are multiple synthesis steps  toward a target molecule P, and the molecule in each step, rd, can be synthesized from hundreds of different reactants. Second, the criteria for a good synthetic route are vague, since different chemists tend to have distinct retrosynthesis approaches toward a target molecule, which largely depend on their personal experience and understanding of retrosynthesis. Moreover, the criteria for a good retrosynthesis route can differ with respect to the chemical scenario. For example, a good retrosynthesis route in industrial manufacturing focuses on stability and cost management, whereas a good route in academia is a new retrosynthesis approach that suffices to tackle a molecule with a complex structure. Furthermore, few reliable retrosynthesis route datasets are open to the public, and researchers tend to use hand-crafted routes to evaluate a retrosynthesis planning algorithm [21] or to compare the generated route with a route reported by chemists via doubleblind A/B tests [22], which is laborious and subjective. Therefore, AI-driven retrosynthesis planning is necessary in order for chemists to accelerate the path-finding process in different scenarios and automate the route evaluation procedure.
toward a target molecule P, and the molecule in each step, rd, can be synthesized from hundreds of different reactants. Second, the criteria for a good synthetic route are vague, since different chemists tend to have distinct retrosynthesis approaches toward a target molecule, which largely depend on their personal experience and understanding of retrosynthesis. Moreover, the criteria for a good retrosynthesis route can differ with respect to the chemical scenario. For example, a good retrosynthesis route in industrial manufacturing focuses on stability and cost management, whereas a good route in academia is a new retrosynthesis approach that suffices to tackle a molecule with a complex structure. Furthermore, few reliable retrosynthesis route datasets are open to the public, and researchers tend to use hand-crafted routes to evaluate a retrosynthesis planning algorithm [21] or to compare the generated route with a route reported by chemists via doubleblind A/B tests [22], which is laborious and subjective. Therefore, AI-driven retrosynthesis planning is necessary in order for chemists to accelerate the path-finding process in different scenarios and automate the route evaluation procedure.
《3. A review of related AI techniques》
3. A review of related AI techniques
《3.1. Sequence-to-sequence (Seq2Seq) models》
3.1. Sequence-to-sequence (Seq2Seq) models
As molecules can be intuitively represented as a sequence, Seq2Seq learning serves as an effective tool for retrosynthesis prediction, such as generating a sequence of reactants from a product. Seq2Seq learning is a type of machine learning approach that has been widely applied to natural language processing (NLP)-related challenges, such as language translation, image caption, and text summarization. It was first proposed in Refs. [23,24] with the encoder–decoder architecture for the machine translation problem: The encoder encodes the whole source sentence into a fixed-length vector, and the decoder generates the target words sequentially. Sutskever et al. [23] and Cho et al. [24] use recurrent neural networks (RNNs) for encoding and decoding, respectively. Considering that RNN-based models cannot capture long-distance dependencies, Bahdanau et al. [25] introduced the attention mechanism [26] to the bidirectional long short-term memory (biLSTM)-based framework, which allows the hidden state to consider the global contextual information. Gehring et al. [27] proposed the convolutional sequence to sequence (ConvS2S) model, which uses multilayer convolution neural networks for encoding and decoding and applies the multi-step attention mechanism to model the global attention for every decoder layer. Vaswani et al. [28] proposed the transformer model, which relies on multi-head attention for encoding and decoding, allowing the model to establish dependencies on arbitrarily distant words in input sentences in a constant number of operations. Thanks to the transformer model’s superior performance, transformer-based methods have become the mainstream, and many extensions have since been proposed [29,30].
《3.2. Graph neural networks》
3.2. Graph neural networks
In addition to encoding molecules as SMILES strings, it is possible to naturally represent molecules as graphs. The proliferation of graph neural network (GNN) [31] research provides various solutions for learning representations of graph-structured information such as molecules, upon which molecular transformations within a reaction can be predicted. Sperduti and Starita [32] were the first to apply neural networks to directed acyclic graphs.
Researchers have also proposed recurrent graph neural networks (RecGNNs), in which the neighbour information iteratively propagates to update the representation of the target node [33,34]. Encouraged by the tremendous success of convolutional neural networks (CNNs) in the computer vision field, Henaff et al. [35] proposed the convolutional graph neural network (ConvGNN). Previous ConvGNNs are with two major categories: spectral-based and spatial-based approaches. Previous ConvGNNs are with two major categories according to different manners of defining graph convolutions: spectral-based approaches follow graph signal processing to adopt filters [36,37], and spatial-based approaches rely on information propagation [38,39]. Later, the graph convolutional network (GCN) [40] was developed to bridge the gap between these approaches. Recently, GCNs have continued to rapidly develop due to their efficiency, flexibility, and generality. With the widespread application of attention mechanisms, some methods adopt an attention mechanism as a node aggregation mechanism, such as graph attention networks (GATs) [41] and gated attention networks (GAANs) [42]. On the basis of these works, many alternative GNN approaches have been developed, including graph auto-encoders (GAEs) [43], graph generative networks (GGNs) [44], and spatial–temporal graph convolutional networks (STGCNs) [45].
《3.3. Search algorithms》
3.3. Search algorithms
Search algorithms retrieve the information stored in a data structure or calculated in a search space, upon which multi-step retrosynthesis prediction relies to plan a synthesis route. In general, search algorithms can be categorized into uninformed searches and informed searches. A uninformed search does not exploit any information about the cost of state transitions; typical examples include depth-first searches and breadth-first searches. In contrast, an informed search contains the heuristic function to evaluate the distance between the current state and the goal state in order to guide the search progress. This guarantees a good solution with a reasonable search time, although the solution is not necessarily optimal. Best-first searches are typical heuristic searches with the concept of a priority queue [46]. The OPEN list stores the currently traversable nodes, and the CLOSED list stores the traversed nodes. Beam search improves the best–first search by expanding the most promising nodes in a limited set [47]. A* search combines the highlights of uniform cost search and best–first search, ensuring the optimal solution [48]. The cost of each state, in this case, is the combination of the actual cost from the starting state to the current state and the heuristic cost from the current state to the goal state. Monte Carlo Tree Search (MCTS) [49] improves the value estimates from the current state to the goal state with a Monte Carlo simulation and consists of four steps: selection, expansion, simulation, and backpropagation. AlphaGo [50] is one of the most famous use cases of MCTS, in which an MCTS searches for possible moves and tracks the results in a search tree of Go.
《3.4. Deep reinforcement learning》
3.4. Deep reinforcement learning
Reinforcement learning (RL) [51] is a machine learning paradigm, in which the agent interacts with the environment and maximizes the notion of cumulative reward with trial and error. RL does not require large-scale annotated datasets and is qualified for sequential decision-making problems with good continuity and innovation. Recently, RL has advanced along with the rapid development of deep learning in learning representations. Deep Q-networks (DQNs) [52], in which convolutional neural networks are used to estimate the Q-value, are a pioneering work in the field of deep RL. Since their development, researchers have proposed several extensions of value-based methods [53]; moreover, model-based [54] and policy gradient methods [55,56] have been proposed instead, in order to predict the after-taking-action state with a predictive model and to directly optimize the policy network. Deep RL also works on more complex decision-making problems, such as those with goal conditions [57], hierarchical task decomposition [58], and multiple agents [59]. Deep RL has achieved wide success in applications such as games [60], robotics [61], autonomous driving [62], and molecule generation [63]. It is believed to be a significant step toward general AI [64].
《4. How AI reshapes retrosynthesis》
4. How AI reshapes retrosynthesis
Unprecedented development in AI over the past decade has led to a proliferation of recent studies that resort to AI techniques to address the challenges discussed in Section 2.2 and improve the performance of retrosynthesis prediction. In Fig. 5, we provide a breakdown of the current landscape of AI-driven retrosynthesis prediction, covering the two major components of single-step retrosynthesis prediction and multi-step retrosynthesis prediction, as well as the two algorithmic design elements of molecular representation and candidate reaction evaluation, which provide the groundwork for single-step and multi-step retrosynthesis prediction, respectively. Together, these four elements establish a design space for AI-driven retrosynthesis prediction methods that orients the development of new algorithms and customization for specific applications. In this section, we break down the existing literature according to the proposed landscape.
《Fig. 5》

Fig. 5. Overview of the retrosynthesis prediction landscape covering the two key components of a retrosynthesis planning algorithm. Single-step retrosynthesis prediction and multi-step retrosynthesis prediction are shown on the right, together with the two indispensable design elements that prepare for these two components: molecular/ reaction representation and candidate reaction evaluation, which are shown on the left.
《4.1. Molecular/reaction representation》
4.1. Molecular/reaction representation
4.1.1. Molecular representation
The mainstream molecular representation methods applied in the retrosynthesis problem include SMILES strings, fingerprints, and graphs, as summarized in Fig. 6. SMILES is the most widely adopted chemical notation system for molecular structure representation [16]. It is easily accessible to chemists, yet interactive enough to allow computers to generate unique line notations. Similar to natural language, the SMILES system combines characters and grammatical rules to specify rigorous structures based on chemical principles [65]. Once a SMILES representation has been created, therefore, researchers can intuitively predict singlestep precursors for retrosynthesis by means of Seq2Seq machine translation models.
The molecular fingerprint is another useful cheminformatics tool to represent molecules [66]. Its core idea is to map a molecule into a bit string or a numeric array of length  where each bit encodes whether or not the molecule contains a unique substructural feature. Consequently, several types of molecular fingerprints exist according to the nature of substructural features, ranging from substructure key-based [67] and path-based [67] features to two-dimensional (2D) circular [68] and three-dimensional (3D) circular [69] features. Cereto-Massagué et al. [67] and Zagidullin et al. [70] present a comparison of the effectiveness of not only these molecular fingerprints but also recently developed learning-based fingerprints. Despite the availability of cheminformatics tools that generate strings or vectors, researchers have proposed representing molecules geometrically. Due to the better preservation of molecular structure characteristics by graph representations [71] and recent development in the field of GNNs, graph molecular representation has been attracting a considerable amount of interest for molecule generation and retrosynthesis prediction.
where each bit encodes whether or not the molecule contains a unique substructural feature. Consequently, several types of molecular fingerprints exist according to the nature of substructural features, ranging from substructure key-based [67] and path-based [67] features to two-dimensional (2D) circular [68] and three-dimensional (3D) circular [69] features. Cereto-Massagué et al. [67] and Zagidullin et al. [70] present a comparison of the effectiveness of not only these molecular fingerprints but also recently developed learning-based fingerprints. Despite the availability of cheminformatics tools that generate strings or vectors, researchers have proposed representing molecules geometrically. Due to the better preservation of molecular structure characteristics by graph representations [71] and recent development in the field of GNNs, graph molecular representation has been attracting a considerable amount of interest for molecule generation and retrosynthesis prediction.
《Fig. 6》

Fig. 6. Summary of three mainstream molecular representation methods used for learning: a SMILES string, a Morgan fingerprint vector, and a graph.
4.1.2. Reaction representation
Chemists define a chemical reaction as a transformation from reactants to products under the condition of reagents. Consequently, a straightforward method of representing reactions is to join strings of reactants, reagents, and products. A reaction SMILES string consists of SMILE strings of reactants, reagents, and products, as well as a ‘‘>” symbol that connects them to signify the reaction direction [16,65]; for example, ‘‘reactants > reagents > products.” Similarly, molecular fingerprints can be concatenated together as a reaction representation. In addition to representing a reaction with molecular representations in a compositional fashion, extracting reaction embeddings from pretrained models is another promising way [72]. Another alternative is to put a chemical reaction into a single condensed graph of reaction (CGR), which is a superposition of the reactant and product graphs [73]. In a CGR, the changes in properties of atoms and bonds within a reaction are highlighted. It is notable that the CGR representation of a reaction relies on atom-to-atom mappings between the reactants and products, and CGRTools [74] is an excellent tool for CGR representation.
《4.2. Single-step retrosynthesis prediction》
4.2. Single-step retrosynthesis prediction
4.2.1. Template selection
A template, as defined in Fig. 2, encodes the changes in connectivity between the products and reactants, while a template set contains a group of templates for different chemical rules. Given a template and a matched product, it is easy to obtain all the reactants in the reaction. Thus, template-based methods [10,11,75,76] have been prescribed for single-step retrosynthesis prediction.
4.2.1.1. Template extraction. The earliest chemical reaction rules were codified by chemists. Experts formulated which reactions were allowed [77]. Since then, hand-crafted rules for generic transformations have been used in retrosynthesis prediction and have achieved great success for several complex products [6,78,79]. With the explosive growth of reactions, there is an urgent need for automatic template extraction, so heuristic algorithms were proposed to build generalized rules from known reaction examples [80–83]. The key idea is to extract the reaction center and include varying numbers of neighboring atoms. The number of neighboring atoms is determined by either a fixed distance to the reaction center or a heuristic of which atoms are relevant. Different numbers dictate various generalization levels of templates. In order to handle stereochemical templates, Coley et al. [84] proposed RDChiral, an RDKit [85] wrapper, for retrosynthesis template extraction.
Rules defined by experts are precise, but there are too few to cover a sufficient fraction of reaction types. Although it is highly efficient to create templates via automatic template extraction, such templates suffer a tradeoff between generalization and correctness. Moreover, the number of templates is huge [84] and continues to increase [86], as novel reactions are discovered continuously.
4.2.1.2. Template retrieval. After extracting templates, another research question remains: how to retrieve a template from the template library, given a target product. The approach of RetroSim [11] mimics a possible decision-making process of a chemist during retrosynthesis prediction; that is, it imitates how similar molecules have been synthesized. Templates that encode the synthetic routes of molecules whose structural motifs are similar to the target molecule are prioritized, and only 100 templates whose products have the greatest similarity to the target molecule  are retrieved. In regard to the similarity measure
are retrieved. In regard to the similarity measure  between molecules, RetroSim considers the 2D structure similarity, which is implemented through two steps. First, a molecule is represented as a vector using Morgan circular fingerprints [69], in which the hyperparameter of the radius refers to the largest size of neighborhood to be considered. The features of each atom [87] are also included. Second, several similarity metrics, including Dice [88], Tanimoto [89], and Tversky [90] similarity, are investigated. It should be noted that the representations and similarity metrics are all implemented in the RDKit software package [85].
between molecules, RetroSim considers the 2D structure similarity, which is implemented through two steps. First, a molecule is represented as a vector using Morgan circular fingerprints [69], in which the hyperparameter of the radius refers to the largest size of neighborhood to be considered. The features of each atom [87] are also included. Second, several similarity metrics, including Dice [88], Tanimoto [89], and Tversky [90] similarity, are investigated. It should be noted that the representations and similarity metrics are all implemented in the RDKit software package [85].
Instead of hinging on explicit molecule similarities, NeuralSym [76,91] casts the problem of template retrieval as a multi-class classification problem in order to automatically learn the patterns of a target molecule, which dictate which template to use. A fully connected neural network is used to perform multi-class classification, as shown in Fig. 7. The input to the neural network is a target molecule represented using Morgan fingerprints [69], and the output nodes correspond to all templates. When provided with a novel target molecule, the well-trained neural network retrieves the top-k templates whose softmax probabilities are the k highest.
《Fig. 7》

Fig. 7. Illustration of the neural-symbolic (NeuralSym) model.
Although NeuralSym is trained to learn a single template given a molecule in benchmark datasets, many potentially useful transformations outside the benchmark are excluded [75]. To address this issue, Fortunato et al. [75] proposed augmenting the benchmark with more synthetic data on template applicability and pretraining a template-relevance network that learns all templates that potentially lead to the reactants. The pretrained templaterelevance network, which was later fine-tuned on real reactions, achieves higher recall in template applicability and more diversity in generated reactants than the previous methods like NeuralSym. Another alternative named MHNreact [92] tackles this issue by encoding both templates and products as input, in contrast to previous approaches, which predict a fixed set of templates when given the target molecule as input. MHNreact then resorts to modern Hopfield networks (MHNs) to learn the association between templates and products.
One obvious limitation of NeuralSym is that the model size grows with the number of templates. To overcome this limitation, Dai et al. [10] proposed the conditional graph logic network (GLN). The GLN directly models and maximizes the conditional joint probability of both templates and reactants using their learned embeddings, which is defined as  where
where  here represents the conditional probability. As for template retrieval by the probability
here represents the conditional probability. As for template retrieval by the probability  , the GLN first screens out all templates matching the target product by evaluating whether the subgraph pattern of a template’s product pT matches a subgraph of the product P. Among these templates, the GLN proceeds to score potential reaction centers within the target project and evaluate the affinity between reactant subgraphs in a template
, the GLN first screens out all templates matching the target product by evaluating whether the subgraph pattern of a template’s product pT matches a subgraph of the product P. Among these templates, the GLN proceeds to score potential reaction centers within the target project and evaluate the affinity between reactant subgraphs in a template  and those in the target product P. The matching scores are calculated via the inner product between molecule embeddings, where each molecule G is embedded as g(G) by the GNN algorithm structure2vec [93]. Finally, the GLN selects the top-k templates with the highest matching scores. The GLN retrieves templates by the similarity computed with GNN embeddings, so that its model size is fixed based on whatever the number of templates is. Nevertheless, the performance of a GLN is still limited by the quality and quantity of templates.
and those in the target product P. The matching scores are calculated via the inner product between molecule embeddings, where each molecule G is embedded as g(G) by the GNN algorithm structure2vec [93]. Finally, the GLN selects the top-k templates with the highest matching scores. The GLN retrieves templates by the similarity computed with GNN embeddings, so that its model size is fixed based on whatever the number of templates is. Nevertheless, the performance of a GLN is still limited by the quality and quantity of templates.
4.2.2. Reaction center prediction
4.2.2.1. Template-based algorithms. Template-based algorithms simply apply the retrieved templates and generate reactants. Retrieved templates provide several candidate reaction centers. RetroSim [11] and NeuralSym [76] pinpoint the reaction center based on the rank of templates, while GLNs [10] predict the reaction center based on the rank of reactants, as defined above.
4.2.2.2. Template-free algorithms. Despite their interpretability and substantial progress, template-based algorithms requiring subgraph isomorphism still suffer from poor scalability and poor generalization to novel reactions. Recently, a considerable literature has grown up around the theme of bypassing templates. Template-free algorithms use machine learning models to straightforwardly predict the reaction centers.
The problem of reaction center identification comes down to link prediction—that is, to predicting whether each edge of a molecule graph  should be disconnected. Following the state-of-the-art practice of link prediction, in which GNNs are used to learn effective representations of each link, Shi et al. [17] adopted the relational GCN (R-GCN) [94] and Yan et al. [8] proposed the edge-enhanced graph attention network (EGAT). A binary classifier is trained to predict whether a bond should be disconnected based on the graphical representation of the bond. It is worth noting that Yan et al. [8] introduced an auxiliary task—that is, predicting the total number of disconnection edges for a target molecule to further improve reaction center identification. In contrast, GraphRetro [95] predicts the potential bond and atom edits simultaneously via a message passing network (MPN) [96]. Atom edits characterize how the number of hydrogens attached to the atom changes from the products to the reactants, which has been not considered by previous approaches [8,17]. Breaking a target molecule at the predicted disconnection bonds leads to a set of intermediate synthons
should be disconnected. Following the state-of-the-art practice of link prediction, in which GNNs are used to learn effective representations of each link, Shi et al. [17] adopted the relational GCN (R-GCN) [94] and Yan et al. [8] proposed the edge-enhanced graph attention network (EGAT). A binary classifier is trained to predict whether a bond should be disconnected based on the graphical representation of the bond. It is worth noting that Yan et al. [8] introduced an auxiliary task—that is, predicting the total number of disconnection edges for a target molecule to further improve reaction center identification. In contrast, GraphRetro [95] predicts the potential bond and atom edits simultaneously via a message passing network (MPN) [96]. Atom edits characterize how the number of hydrogens attached to the atom changes from the products to the reactants, which has been not considered by previous approaches [8,17]. Breaking a target molecule at the predicted disconnection bonds leads to a set of intermediate synthons  , which lay a solid foundation for the generation of valid reactants.
, which lay a solid foundation for the generation of valid reactants.
4.2.3. Reactant generation
4.2.3.1. Template-based generation. Reactants are derived by subgraph matching, given a target product and a template [84]. RetroSim [11] and NeuralSym [76] sort the templates by their scores and generate the top-k reactants following the same order of top-k templates. GLNs [10] rank all reactants after the selected template is applied to the target molecule. The ranking score of the reactants  is calculated by the inner product between the embedding of the target molecule P and the mean embedding of all reactants
is calculated by the inner product between the embedding of the target molecule P and the mean embedding of all reactants  , given that ① pT is a subgraph of P, ② the size of the reactant set is equal to that of reactant subgraphs
, given that ① pT is a subgraph of P, ② the size of the reactant set is equal to that of reactant subgraphs  of the template T, and ③ each reactant subgraph of T is a subgraph of at least one reactant in under a permutation π(·). Given a novel target molecule, the predicted reactants are sampled from the joint probability of the matched templates and generated reactants using a beam search of size k.
of the template T, and ③ each reactant subgraph of T is a subgraph of at least one reactant in under a permutation π(·). Given a novel target molecule, the predicted reactants are sampled from the joint probability of the matched templates and generated reactants using a beam search of size k.
4.2.3.2. Synthon-based generation. In G2Gs [17] and RetroExpert [8], a set of intermediate synthons  must be transformed into chemically valid reactants
must be transformed into chemically valid reactants  . A transformer [28] is adopted in Ref. [8] to translate synthons into reactants. To ensure that the translation model is invariant of the order of synthons, Yan et al. [8] proposed augmenting the training pairs by shuffling the order of synthons and reactants. In Ref. [17], however, the researchers formulated the translation problem as a variational graph translation. Conditioned on the synthons, the generation of the reactant graph consists of multiple steps of graph transformation actions. More specifically, G2Gs considers four consecutive actions at each step: ① predicting the termination of graph translation, ② predicting the type of the added atom, ③ predicting the other atom to connect with, and ④ predicting the type of bond between the two atoms. GraphRetro [95] directly learns an MPN [96] to select the leaving groups that should be attached to synthons in order to generate complete reactants. It should be noted that the leaving groups come from a preprocessed discrete vocabulary. The selected leaving groups are later attached to synthons by adding a bond Fig. 7. Illustration of the neural-symbolic (NeuralSym) model. between them, in accordance with the valency constraint.
. A transformer [28] is adopted in Ref. [8] to translate synthons into reactants. To ensure that the translation model is invariant of the order of synthons, Yan et al. [8] proposed augmenting the training pairs by shuffling the order of synthons and reactants. In Ref. [17], however, the researchers formulated the translation problem as a variational graph translation. Conditioned on the synthons, the generation of the reactant graph consists of multiple steps of graph transformation actions. More specifically, G2Gs considers four consecutive actions at each step: ① predicting the termination of graph translation, ② predicting the type of the added atom, ③ predicting the other atom to connect with, and ④ predicting the type of bond between the two atoms. GraphRetro [95] directly learns an MPN [96] to select the leaving groups that should be attached to synthons in order to generate complete reactants. It should be noted that the leaving groups come from a preprocessed discrete vocabulary. The selected leaving groups are later attached to synthons by adding a bond Fig. 7. Illustration of the neural-symbolic (NeuralSym) model. between them, in accordance with the valency constraint.
4.2.3.3. Direct generation. Molecule edit graph attention network (MEGAN) [97] generates reactants directly from graph representations of the target molecule P. MEGAN first embeds the target product with GNNs, and then modifies the target product gradually by means of a transformer-based [28] decoder. Analogous to G2Gs [17], MEGAN chooses one action at each step, generating intermediate substrates or an end substrate. Candidate actions are defined as follows: ① editing atom properties, including changing chirality, aromaticity, and so forth; ② editing the bond between two atoms, which involves adding, deleting, or editing the bond; ③ adding a new atom to the graph, where both the atom and its connection with one of the existing atoms are added; ④ adding a new benzene ring to the graph, where a complete benzene ring is appended to a selected carbon atom; and ⑤ stopping generation, which is the end of the generation process. As the reaction center is unclear, the model is trained by maximizing the log-likelihood objective with a fixed ordering of actions [98]. Table 1 [97] lists the action priorities defined in MEGAN. Without dissociating the reaction center prediction from the reactant prediction, MEGAN enjoys the advantage of end-to-end training.
Aside from GNNs, other researchers have formulated singlestep retrosynthesis prediction as an Seq2Seq machine translation problem, where the input is the SMILES string [16] of a target molecule and the output is a sequence of SMILES for all reactants. As shown in Fig. 8, Liu et al. [7] adopted biLSTM [99] as the neural Seq2Seq model. To leverage more recent advances in neural machine translation and improve the retrosynthesis prediction accuracy, Karpov et al. [100] replaced biLSTM with the transformer model [28].
《Table 1》
Table 1 Action prioritization used for training MEGAN [97].

The main weakness of Seq2Seq translation algorithms is their unsatisfactory performance, which is due to three reasons: First, the representation of molecules as strings ignores the rich chemical structures and interplay between atoms. Second, the chemical validity of generated SMILES strings is not guaranteed. Third, the end-to-end training strategy fails to include any chemistry knowledge within abundant reactions. In addition to their poor performance, Seq2Seq models lack interpretability regarding their predictions. On the other hand, direct generation enjoys the following exclusive benefits: ① Although it is challenging for template-based algorithms to generate reagents, including solvents, catalysts, and other substances, to lead to a reaction, it is possible for direct generation; and ② direct generation tends to produce more diverse reactions, which is crucial for multi-step retrosynthesis, in comparison with synthon-based generation. In future, there is hope that the performance of Seq2Seq methods can be improved via data augmentation and enrichment. Recently, dual template-based proposing (DualTB) [101] has made progress in providing a unified view of both template-free methods and template-based methods by formulating a novel energy-based framework. DualTB easily accommodates the two types of methods by defining different energy functions; during the inference phase, reactant candidates obtained by template matching or generated by the model are ranked according to the predicted energy score.
《Fig. 8》

Fig. 8. Illustration of the biLSTM model for retrosynthesis prediction.
《4.3. Candidate reaction evaluation》
4.3. Candidate reaction evaluation
4.3.1. Feasibility
4.3.1.1. Retrosynthesis scoring. The retrosynthesis model itself is capable of ranking the predicted reactions with different scores. For example, RetroSim [11] uses molecular similarity to rank candidate chemical reactions; the overall similarityscores) are the product of the product similarity (sprod) and the reactant similarity (sreac), that is, s = sprod ×sreac. A GLN [10] decomposes the scoring of candidate reactions into two parts: the template score function (ω1) and the reactant score function (ω2). The template score function is further decomposed into two parts: the product score (v1) and the reactant score (v2). Together, these scores dictate the ranking of a predicted reaction. For synthon-based and direct generation methods [8,100], the score amounts to the log probability of the whole SMILES string, which is the sum of the log probabilities for all predicted tokens. This ranking of candidate reactions is the same as that is performed by a beam search.
4.3.1.2. Round-trip prediction. A rigorous evaluation of the feasibility of a predicted retrosynthesis would be to investigate whether the resulting precursors (i.e., reactants) can generate the product as expected, which would require a forward chemical reaction prediction model. Schwaller et al. [15] coined the term ‘‘round-trip evaluation” to describe such an evaluation and introduced two round-trip evaluation metrics: round-trip accuracy and coverage. Round-trip accuracy refers to the ratio of valid reactants to all reactants predicted by the backward chemical reaction prediction model, where ‘‘valid” means that the reactants can produce the target product, as predicted by the forward chemical reaction prediction model. Coverage evaluates the number of target products for which the retrosynthetic model produces at least one valid set of reactants. The coverage evaluation metric encourages a retrosynthesis model to produce valid reactants for a wide variety of target molecules and thereby makes up for the shortcoming of round-trip accuracy, which may over-reward models that produce many valid precursors for only a few reactions.
4.3.2. Diversity
In the chemical reaction space, there are many ways of generating a molecule, and different ways may correspond to different reaction types. Here, it should be noted that organic reactions with the same reaction mechanism belong in the same category. A good retrosynthesis model is expected to produce as diverse synthetic routes as possible for the target molecule, where the diversity can be evaluated by the Jensen–Shannon divergence (JSD) [15] between the likelihood distributions of a reaction belonging to N reaction types. The JSD is computed as follows:

where PDi denotes the ith probability distribution and H(PDi) denotes the Shannon entropy for the distribution PDi.
4.3.2.1. Rule-based classification. Reactions have conventionally been classified manually, based on the class of the product, the functional group, the reagent used, or even the inventor of the reaction. However, the coverage of reactions using such classification methods is far from enough. Model-based methods were later proposed, which rely on predefined definitions of the reaction center and consider the bonds that change during the reaction [102]. As a means of overcoming the limitation of model-based methods that overlook the functionality and subclasses beyond the reaction center, the data-driven approach [103] identifies multiple levels of description of the reaction center by extending the reaction center to include the neighboring bonds. The hash code—also known as the reaction type—is automatically calculated for each level of description of the reaction center, taking into account the atom type, valence state, total number of bonded hydrogen atoms, number of p-electrons, aromaticity, formal charges, and reaction center information. Another data-driven method [104] groups reactions that share the reaction classification (RC) number into one category, where the RC number characterizes the conversion patterns of the types of atom in the reaction center. The condensed reaction graph (CRG) approach [73] directly merges all of the reactants and products of a reaction into an imaginary transition state or pseudomolecule, so that the descriptors of pseudo-molecules can be used to evaluate the similarity between reactions. Data-driven approaches, however, are likely to fail to detect reactions that have a similar underlying mechanism but different topology of the extended reaction center.
Using a large rule base of known reaction mechanisms or transformations, NameRXN [105] is capable of categorizing roughly 1000 name reactions. Although it is detailed and accurate, NameRXN can usually identify the reaction classes of only about 50% of reactions. As a result, it is necessary to use learning-based classification algorithms to generalize to the remaining unclassified reactions.
4.3.2.2. Learning-based classification. The increasing availability of large reaction datasets has promoted the development of machine learning algorithms to classify chemical reactions, where a classifier is directly trained with a set of classified reactions by NameRXN. Schneider et al. [106] investigated a range of reactiondifference fingerprints as features and five machine learning models, including random forest, naïve Bayes, k-means, logistic regression, and k-nearest neighbors, for classifying 50 reaction types in the name reaction ontology (RXNO) [103]. A hierarchical classification model was later proposed in Ref. [107], in which the conformal prediction (CP) framework is used to predict 336 reaction types in a hierarchy with confidence measures. Recently, Schwaller et al. [72] used a transformer-based model named RXNFP to achieve a state-of-the-art classification accuracy of 98.2%, using only the SMILES representations of chemical reactions as input and removing the need for the automated annotation of reaction centers.
4.3.3. Efficiency
Another desirable quality for evaluating whether a reaction should be prioritized is efficiency, which is measured using the synthetic complexity score of the reaction’s reactants and the yield of the target products. Reactions with reactants that are easier to synthesize and a higher yield of the target product are preferred.
4.3.3.1. SCScore. Coley et al. [108] proposed a method to calculate the synthetic complexity of a reactant. The key ideas behind the SCScore are twofold: First, if a molecule appears to require many reaction steps to be synthesized by traditional methods, it is considered to be difficult to make and is assigned a higher SCScore; second, the more frequently a molecule appears as a reactant, the lower its synthesis complexity tends to be. It should be noted that the SCScore of a reactant is always less than or equal to that of the product.
4.3.3.2. Yield. In a chemical reaction, it is possible that some of the reactants will not react to give a product. The yield of a chemical reaction describes the percentage of the reactants that are successfully converted into the product, relative to the theoretical maximum:

where y denotes the yield of a chemical reaction, Va is the actual production quantity of the target product, and Vt is the theoretical production quantity.
Recently, there has been increasing interest in predicting the yield using machine learning methods. Based on a reaction representation by means of molecular fingerprints and chemicallinguistic descriptors (CLDs) [109], Skoraczyn´ ski et al. [110] compared a variety of machine learning methods (e.g., decision tree and random forest) for predicting the reaction yield. Inspired by the recent success of the transformer-based model in the classification of reaction types [72], Schwaller et al. [111] fine-tuned the RXNFP model [72] to predict the yield. Unfortunately, the performance was not very satisfactory, as the yield prediction problem presents the challenge of ill-defined and noisy annotations.
《4.4. Multi-step retrosynthesis prediction》
4.4. Multi-step retrosynthesis prediction
When working toward multi-step retrosynthesis planning, the agent needs to choose a reaction from the legal candidate set, given a target molecule at each step, observe the feedback, and then make the next selection move on a new target molecule. This process bears a striking similarity to the sequential decision-making in a general strategic decision-making game; in other words, the problem of multi-step retrosynthesis planning can be framed as a one-player game [112]. Therefore, two central research issues that must be addressed in multi-step retrosynthesis planning are the same as those in solving a game—namely, how to efficiently search for a successful route and how to estimate the value of each route. Moreover, unlike games such as chess, where the winning state is binary, retrosynthesis planning requires the winning state to be differentiated according to the cost of synthesis. The third daunting research challenge that remains is to evaluate the synthetic cost, which involves multiple quantities such as the number of synthesis steps, the yield, and the total cost of the commercially available reactants.
In the next section, we review the recent methods summarized in Table 2 [15,21,22,112–115] and take a closer look at how they learn a prior value estimation neural network in an offline manner (offline learning), how they perform an online search in which the estimated value can be optionally refined (online search), and how they evaluate a retrosynthesis route (evaluation).
《Table 2》
Table 2 Summary of existing methods for multi-step retrosynthesis planning algorithms with regard to the two central research issues.

PNS: proof-number search; 3N-MCTS: MCTS with three neural networks; DFPN-E: depth-first proof-number search with heuristic edge initialization; ASICS: advanced system for intelligence chemical synthesis.
4.4.1. Offline learning
Due to the difficulty in heuristically and accurately determining the retrosynthesis value (i.e., cost) of a molecule, researchers are motivated to learn the value from historically simulated retrosynthesis planning experiences. A learned policy that is parametrized with a neural network in such an offline manner serves as a reliable prior probability for a search algorithm. The crux, of course, is to construct a training dataset of retrosynthesis planning routes, where each molecule is annotated with the defined cost of its synthetic route. Existing works rely on routes either designed by chemists or hand-crafted with a single-step reaction dataset; as a result, established retrosynthesis route datasets suffer from limited size, noise, and imbalance due to being based on only successful routes. Notably, Segler at al. [22] used reactions from the literature as the ground-truth to pretrain a neural network that determines the rank of reaction templates for generating a retrosynthesis reaction when given a specific molecule. A policy network was later incorporated into the upper confidence bound (UCB) formula in order to balance the exploration and exploitation in an MCTS.
In contrast, instead of directly using reactions from the literature, Chen et al. [21] hand-crafted a dataset of retrosynthesis routes for pretraining the policy network. They analyzed each molecule that appeared inside a single-step reaction dataset by checking whether the molecule could be synthesized by existing reactions within the dataset and connecting the shortest route to synthesize the molecule. All the routes were collected in order to pretrain the policy network estimating the value of synthesizing a molecule, which was used as the heuristic in an A* search. Schwaller et al. [15] use pretrained reaction complexity metrics, such as SCScore [108], as heuristics to form the Bayesian-like probability as the exploration prior knowledge.
4.4.2. Online searching
The online search is the most important component of retrosynthesis planning. There are many search algorithms in AI, ranging from uninformed algorithms such as greedy depth-first search to informed algorithms such as proof-number search (PNS) and A*. Existing studies on multi-step retrosynthesis prediction favor informed algorithms that are more efficient and accurate, as the use of a heuristic function can potentially result in good solutions. The definition of a heuristic function, however, is non-trivial, as the value of each reaction is uncertain until the whole synthetic route is finished, and the definition of a good synthetic route is vague and subjective. For example, MCTS with three neural networks (3NMCTS) [22] uses the traditional UCB formula as the heuristic in order to attain a balance between exploration and exploitation. In addition to a pretrained policy network, which dictates the exploitation only, 3N-MCTS incorporates the estimated value through its fast rollout policy network to refine the heuristic function in UCB. Fig. 9 demonstrates the four phases of retrosynthesis planning by means of 3N-MCTS, and Fig. 10 provides an exemplar of a returned search tree. HyperGraph search [15] combines the reaction confidence score with the same single-step retrosynthesis model used in 3N-MCTS and SCScore to form a Bayesian-like probability as the heuristic for a beam search. As an alternative, PNS [116] is an effective search algorithm for solving games, and especially for solving difficult endgame positions. By modeling retrosynthesis planning as a two-player game, Heifets and Jurisica [112] directly applied a depth-first proof-number search (DFPN) to multi-step retrosynthesis prediction, based on an AND–OR tree. Kishimoto et al. [114] modified the heuristic to avoid heuristic degeneration in a lopsided search space. Chen et al. [21] combined PNS and an A* heuristic search for retrosynthesis planning, where a pretrained policy network is used to obtain a prior estimate of the retrosynthesis cost of each unsolved molecule. Similarly, Jeong et al. [115] performed a hybrid A*-like search on both a predefined reaction knowledge graph and machine learning-based reaction predictions, where the heuristic was designed to include the molecular similarity, synthetic accessibility score, and likelihood score.
《Fig. 9》

Fig. 9. Illustration of four phases in an MCTS for retrosynthesis planning.
《Fig. 10》

Fig. 10. An exemplar retrosynthesis search tree. The search tree starts from the target molecule; each node in the tree is either discarded or picked up for expansion; a leaf node ends when it has no child node to expand or arrives at the starting materials.
4.4.3. Evaluation
The evaluation of a retrosynthesis planning model can often be done in different, multidimensional ways. Here, we divide the evaluation of a search algorithm into two main metrics: efficiency and quality. In terms of efficiency, Chen et al. [21] used the number of calls of single-step retrosynthesis models as a surrogate for the time required to synthesize. Two commonly adopted methods have been used as a metric of quality: ① evaluating the success rate of an arbitrary set of molecules that finally finish a retrosynthesis pathway (i.e., that arrive at the starting materials) within a fixed number of single-step inference calls; and ② evaluating the cost and length of a route. One approach for evaluating the cost involves calculating the total sum of the negative log-likelihood (NLL) of all single-step reactions along the route. (The length of a route simply amounts to the total number of reactions in a given route.) A hypergraph search [15] introduces chemoselectivity into the route evaluation. Unfortunately, there is no existing automatic tool to quantify the chemoselectivity accuracy of a reaction.
《5. Review of current progress》
5. Review of current progress
《5.1. Single-step retrosynthesis prediction》
5.1. Single-step retrosynthesis prediction
5.1.1. Datasets
Derived from US Patent and Trademark Office (USPTO)-granted patents [117], USPTO-50 K [118] and USPTO-full [119] are two widely adopted benchmark datasets for single-step retrosynthesis prediction. USPTO contains 50 K reactions covering ten reaction types, and USPTO-full consists of 950 K purified reactions from USPTO 1976–2016 without being restricted to specific reaction types. As suggested in Refs. [7,8], both USPTO-50 K and USPTO-full have random training/validation/test splits of 8:1:1.
5.1.2. Architectural and training details
Table 3 [7,8,10,17,76,95,97,100,101] lists the specific architecture and training efficiency for each singe-step retrosynthesis prediction algorithm. The first three template-based methods construct template-retrieval networks, and the next batch of methods (Seq2Seq, transformer, and MEGAN) build a sequence or graph translation model. The last three algorithms are semi-templatebased, as they first build a reaction center prediction network for predicting reaction centers, and then construct a reactant generation network for transforming synthons into reactants.
《Table 3》
Table 3 Summary of the architectural and training details in existing single-step retrosynthesis prediction algorithms.



ReLU: rectified linear unit; ELU: exponential linear unit; GPU: graphics processing unit; MLP: multilayer perceptron.
5.1.3. Evaluation metrics
The top-k exact match accuracy is a common evaluation metric used to evaluate whether the ground-truth reactants fall into the list of top-k predicted reactants. The accuracy is computed by matching the canonical SMILES strings of the predicted reactants with the ground-truth, typically using the RDKit [85] package for canonicalization. It is notable that RDKit is a widely popular open-source cheminformatics toolkit.
Table 4 [7,8,10,11,17,76,95,97,100,101] summarizes the results of template-based and template-free algorithms on USPTO-50 K, where semi-template-based methods refer to two-stage template-free algorithms that first predict the reaction center and then generate reactants. We list the major conclusions as follows: First, the rule-based algorithm, such as ExpertSys, performs poorly. Second, template-based methods, all of which use the same set of templates following Ref. [10], are quite competitive, outperforming the Seq2Seq template-free approaches. Third, upon reaction center identification, the two-stage template-free algorithms tend to be the best performing among template-free algorithms, especially for the top-1 prediction. Finally, the GLN, which maximizes the joint probability of the templates and reactants, is favored for top-5/10/20/50 predictions, if the templates are automatically extracted without much effort. Given that a target molecule in practice is wildly different from those in templates, the semi-template-based methods with both superior generalization ability and competitive predictive capability are preferred. In Table 5 [10,11,76,97], we demonstrate the scalability of four selected algorithms on the USPTO-full dataset. It can be observed that the GLN and DualTB outperform both the similarity-based template-retrieval method RetroSim and the classification-based template-retrieval method NeuralSym.
《Table 4》
Table 4 Comparison of existing algorithms for single-step retrosynthesis prediction on USPTO-50 K.

《Table 5》
Table 5 Comparison of selected algorithms on USPTO-full.

《5.2. Multi-step retrosynthesis prediction》
5.2. Multi-step retrosynthesis prediction
Evaluating the quality of a retrosynthesis planning route is particularly challenging, as discussed in Section 4.4. First, the evaluation metric is vague: The reasons why chemists consider a synthetic route to be good could range from a short pathway or cheap starting materials to easily satisfied reaction conditions, among other options. Second, there is no large-scale benchmark dataset consisting of good synthetic routes that can be used to quantitatively compare the performance of different multi-step retrosynthesis prediction algorithms. Below are some datasets and evaluation metrics as compromise.
5.2.1. Datasets
5.2.1.1. A small-scale benchmark with expert annotation. Due to the difficulty of collecting the synthetic routes that have been demonstrated to be effective in published papers, Heifets and Jurisica [112] constructed a small benchmark including 20 synthesis problems that appeared in the exams of the chemistry course at Massachusetts Institute of Technology. The answers were provided by the instructors. The main disadvantage of this benchmark is its extremely small size.
5.2.1.2. A large-scale benchmark with automatic annotation. Chen et al. [21] constructed a large-scale benchmark dataset of retrosynthesis routes based on the USPTO dataset [117] made up of onestep reactions. A synthetic route for a molecule is automatically constructed by iteratively applying a one-step reaction in USPTO; if multiple routes are found, the shortest one is selected. There are a total of 299 202 training routes, 65 274 validation routes, and 189 test routes. This dataset, however, is disadvantaged by the limited chemical reactions covered by the USPTO and the evaluation of route quality by length.
5.2.1.3. Large-scale benchmarks without annotation. In Refs. [22,114], the authors directly selected 497 diverse molecules first reported in or after 2015 as known starting molecules and 897 target molecules that are not in the set of starting molecules, respectively.
5.2.2. Evaluation metrics
5.2.2.1. Double-blind AB test by chemists. For a target molecule whose synthetic route as specified by experts is provided, chemists are invited to directly evaluate the goodness of a route predicted by an algorithm via the double-blind AB test [22]. They are required to choose one of the two routes based on personal reference and synthetic feasibility.
5.2.2.2. Effectiveness metrics. Different retrosynthesis planning algorithms are compared with each other and with the routes developed by manual or automatic annotation, if any. The effectiveness metrics in the literature include the percentage or number of molecules being solved, the length of a route, and the total cost summarizing the NLL of all reactions along the route. Typically, retrosynthesis planning algorithms take the confidence score for each reaction predicted by a single-step retrosynthesis model as the loglikelihood. On the other hand, Mo et al. [120] introduced an innovative data-driven approach for route evaluation, which proposes a dynamic tree-structured long short-term memory (LSTM) model to encode pathways with various structures into an embedding representation. The embedding consists of the pathway-level information, which is ready for use in both strategic route evaluation and route similarity checking.
5.2.2.3. Efficiency metrics. Aside from the effectiveness, the efficiency of a search algorithm is of major concern. Existing works evaluate the time cost of solving a molecule and the number of nodes expanded.
We have presented the performance comparison of selected algorithms as published elsewhere, although there is no consistent and comprehensive performance comparison of all of the multistep retrosynthesis prediction algorithms that we have introduced. Closer inspection of Table 6 [114] and Table 7 [21]) shows that the search algorithms based on the AND–OR tree, including Retro* and the PNS variant depth-first proof-number search with heuristic edge initialization (DFPN-E), consistently outperform MCTS in terms of the route length and the search runtime. Despite the outstanding performance of Retro* in Table 7 [21], we encourage more comparison on other datasets. The training routes used to train the value estimation neural network of Retro* and the test routes are constructed from the same dataset. It is not surprising, in this case, that Retro* outperforms its competitors. We also recommend more empirical studies of the policy iteration algorithm [113], which improves the value estimation in comparison with Retro*. More advanced attempts could involve taking the best part of both algorithms—that is, estimating the value based on Ref. [113] and searching with A* following Ref. [21].
《Table 6》
Table 6 Performance comparison on 897 target molecules without annotation [114].

《Table 7》
Table 7 Performance comparison on 189 test molecules with automatic annotation [21].

《5.3. Full-fledged retrosynthesis planning frameworks 》
5.3. Full-fledged retrosynthesis planning frameworks
Since 1959, when Corey and Wipke [4] introduced the first software, computer-aided retrosynthesis has been a very active and burgeoning area of research. The pioneers LHASA [121,122] and SECS [123] constantly inspire the development of retrosynthesis. Unfortunately, these two packages and their rule-based followups [6] suffer from two significant challenges: ① Hand-coded rules are of limited size and diversity, as expanding the rule set is very laborious; and ② there is a lack of holistic metrics to rank or score pathways. This has motivated the need for automatically generated reaction rules, with the framework of ARChem Route Designer [83] as a representative. Still, ARChem fails to take stereochemistry and regiochemistry into consideration.
《Table 8》
Table 8 Summary of recent full-fledged retrosynthesis planning frameworks.

GUI: graph user interface; CAS: Chemical Abstracts Service; API: application programming interface.
Recently, the tremendous opportunities offered by deep learning and AI techniques have cultivated several excellent and popular full-fledged retrosynthesis planning frameworks, as summarized in Table 8 [13–15,124–126]. Synthia [13,124], which was previously known as Chematica, resorts to the chemical scoring function and the reaction scoring function to determine which hand-coded rule (among 100 000 hand-coded rules provided by experienced chemists) to apply and which pathway to select. It should be noted that these two functions can be edited by users. Synthia explores the synthetic space by performing a multiple beam-like search. In Ref. [13], Synthia is demonstrated to be capable of proposing experimentally successful pathways for multiple drug-like molecules. Although hand-coded rules or templates are used in Synthia, it is worth noting that the template applicability is further fine-tuned to evaluate site- or regioselectivity by a machine learning model (i.e., a classifier).
Instead of being commercially available, ASKCOS [14] is opensource and more AI-driven compared to previous frameworks. ASKCOS automatically extracts 163 273 templates from the Reaxys database and trains a feedforward neural network to predict which of the templates to apply for a target molecule. For multi-step retrosynthesis planning, ASKCOS adopts a root-parallelized MCTS. To guarantee the plausibility and quality of the returned pathways, ASKCOS removes low-quality ones with the in-scope filter proposed in Ref. [22] and pathways that cannot generate the target product by means of a forward prediction model. The proposed synthesis pathways of 15 medicinal molecules have been finished via a completely automatic robotic system, with easily accessible reagents and reactants.
Another open-source software, AiZynthFinder [125], is similar to ASKCOS on the algorithmic side, but aims to provide a flexible open-source benchmark that supports continuous development on it. RoboRXN [15,126] is pushing ahead with single-step retrosynthesis prediction from template-based methods to the completely template-free Molecular Transformer. More impressively, RoboRXN first introduced four new metrics, including coverage, class diversity, round-trip accuracy, and the JSD, to thoroughly assess the Molecular Transformer; a hypergraph of potential reactants is constructed with the help of these metrics, and a beam search is performed on the hypergraph to conclude a pathway. Researchers from International Business Machines Corporation have demonstrated that RoboRXN can synthesize 3-bromobenzylamine in as little as 1 h, following its own planned pathway. Other frameworks, such as SciFindern and Spaya AI, are commercial and partly undisclosed.
The accessibility of these full-fledged frameworks is largely dictated by the availability of the reaction database or hand-coded reaction rules (templates) and the availability of starting material databases (e.g., eMolecules). The commercial frameworks rely on commercial databases, while open-source ones resort to publicly available databases. Table 8 shows that the interfaces requiring the minimum expertise, such as inputting SMILES and drawing a molecule, are quite user-friendly in order to facilitate the adoption of these retrosynthesis frameworks by chemists. We would expect the current state-of-the-art single-step retrosynthesis prediction algorithms (e.g., DualTB [101]) and state-of-the-art multi-step planning methods (e.g., Retro* [21]) to replace their counterparts in these existing frameworks, especially in the open-source ASKCOS and AiZynthFinder, so that the planned pathways achieve more exciting breakthroughs in either academia or commercial usage.
《6. Conclusion and outlook》
6. Conclusion and outlook
The past three years have seen the rapid development of machine-aided retrosynthesis planning. The pure data-oriented AI approach [22] has made significant advances in both effectiveness and efficiency, outperforming conventional machines based on extracted rules and hand-designed heuristics. The continuously evolving hybrid expert-AI system called Synthia [13,124] complements data-oriented approaches in planning the syntheses of complex targets. The open-source framework ASKCOS [14] even pushes ahead with robotic execution of the planned chemical synthesis routes, further saving the time and effort of expert chemists. This work is intended to provide a comprehensive review of existing retrosynthesis approaches in the algorithm design space, so as to ① inform computer scientists and computational chemists of the explored and remaining research problems in the retrosynthesis of medicinal chemistry targets, ② encourage the integrations or development of state-of-the-art machine learning methods into full-fledged frameworks that provide chemist-friendly interfaces and are still the top choice of chemists, and ③ inspire more research to improve any research aspect in the whole design space, such as reaction representation or reaction center prediction. There is still a long way to go before machines are widely accepted as helpful assistants for chemists. We recommend several possible future research directions.
《6.1. Out-of-distribution generalization and detection》
6.1. Out-of-distribution generalization and detection
The distribution gap between the reaction datasets that are used to train either single-step or multi-step retrosynthesis prediction models and the testing molecules has a non-negligible impact on the retrosynthesis planning performance. As thoroughly investigated in Ref. [127], a smaller overlap between the training reaction dataset and new molecules (reactions)—such as the AstraZeneca virtual libraries that contain general medicinal chemistry targets, as reported in Ref. [127]—will lead to a decreased generalization capacity of AI-driven approaches. Generalizing the template-based methods and AI-driven methods to solve general medicinal chemistry targets out of the distribution of existing reaction datasets and even to process chemistry targets would cause such approaches to be at high risk. Since the goal of retrosynthesis is to synthesize novel molecules, future work on retrosynthesis prediction should stress the establishment of models that are capable of generalizing even to out-of-distribution molecules and reactions with both ① competitive predictive accuracy and ② uncertainty calibration. Such a guarantee in predictive uncertainty would allow appropriate intervention by expert chemists when an out-of-distribution molecule or reaction is known to have a high level of uncertainty. Modular models with compositionality could be a qualified candidate for generalizing to out-ofdistribution samples.
《6.2. Route evaluation》
6.2. Route evaluation
With the development of computer-aided synthesis planning (CASP), automatic route evaluation becomes urgent when evaluating the performance of different CASP programs. In addition to considering the quality of each single-step reaction inside a route, the overall quality of a route dictated by a retrosynthesis strategy is essential. For example, a convergent synthesis is a priority for a retrosynthesis strategy, since it reduces the maximum length of the retrosynthesis pathway and improves the overall yield rate; protection and de-protection reaction types appearing together inside a single retrosynthesis route contribute to avoiding non-selective reactions. Although designing different heuristics in the existing literature can guide the CASP program to follow a certain strategic type of retrosynthesis planning, it also weakens the potential of CASP in finding an overall ‘‘smarter” path. Future work could apply the data-driven approach, while also providing a holistic route evaluation with a better encoding of pathway-level information.
《6.3. Knowledge graphs and reasoning》
6.3. Knowledge graphs and reasoning
Knowledge graphs are semantic and structured representations of human knowledge, which greatly facilitate many informationprocessing and natural language-understanding problems [128,129]. Analogously, in the field of retrosynthesis, constructing a knowledge graph of molecules and the relationships connecting the molecules would effectively complement the existing unstructured and neural retrosynthesis algorithms. First, the retrosynthetic route of the target molecule could be regularized by the synthesis route of the most similar molecule queried in the knowledge graph. Second, it is much more efficient to incorporate a new reaction or novel synthetic route annotated by chemists into an online knowledge graph than to take it as a training example for updating a neural retrosynthesis algorithm. To achieve this goal, we argue the critical importance of ① defining categories of molecules by the type of reaction that synthesizes the molecules; ② building on such categories to formulate various categories of relationships between molecules, such as ‘‘Category.Share,” ‘‘ReactantOf,” and ‘‘ReagentOf;” and ③ learning the contextual representation of molecules by simultaneously considering the individuality and connection of other molecules to reduce the noise and ambiguity. Jeong et al. [115] has already made a reasonable attempt toward the first research challenge above by exploring the use of a knowledge graph to regularize predicted reactions. The remaining challenges—that is, how to continually update the knowledge graph and learn contextual representations of molecules for facilitating retrosynthesis prediction and planning—are open to be explored next.
《Acknowledgments》
Acknowledgments
The authors are thankful for financial support from the Tencent AI Lab Rhino-Bird Gift Fund (9229073), the Project by Shanghai Artificial Intelligence Laboratory (P22KS00111), and the Starry Night Science Fund of Zhejiang University Shanghai Institute for Advanced Study.
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Yinjie Jiang, Yemin Yu, Ming Kong, Yu Mei, Luotian Yuan, Zhengxing Huang, Zhihua Wang, Huaxiu Yao, James Zou, Connor W. Coley, and Ying Wei declare that they have no conflict of interest or financial conflicts to disclose.











 京公网安备 11010502051620号
京公网安备 11010502051620号




