

《1. Introduction》
1. Introduction
Moxifloxacin is a fourth-generation fluoroquinolone antibiotic with broad-spectrum activity that is particularly effective against infectious bacterial diseases [1]. Compared to other routes, the lipase-catalyzed biosynthetic route of a significant chiral intermediate is favored for the industrial production of moxifloxacin (S,S)-2,8-diazobicyclo[4.3.0]nonane from racemic cis-(±)-dimethyl 1-acetylpiperidine-2,3-dicarboxylate (cis-(±)-1) (Fig. 1) [2–4]. Numerous lipases (E.C.3.1.1.3) with a wide substrate spectrum are available on the market and in academia, such as Novozym® 435 or the well-known Candida antarctica lipase B (CALB); however, these lipases are limited in their activity and stereoselectivity toward specific substrates, in this case cis-(±)-1, ultimately resulting in low stereoselectivity, large enzyme usage, and increased production costs [5,6]. Thus, highly stereoselective and efficient lipases are highly desirable for this biotransformation.
《Fig. 1》
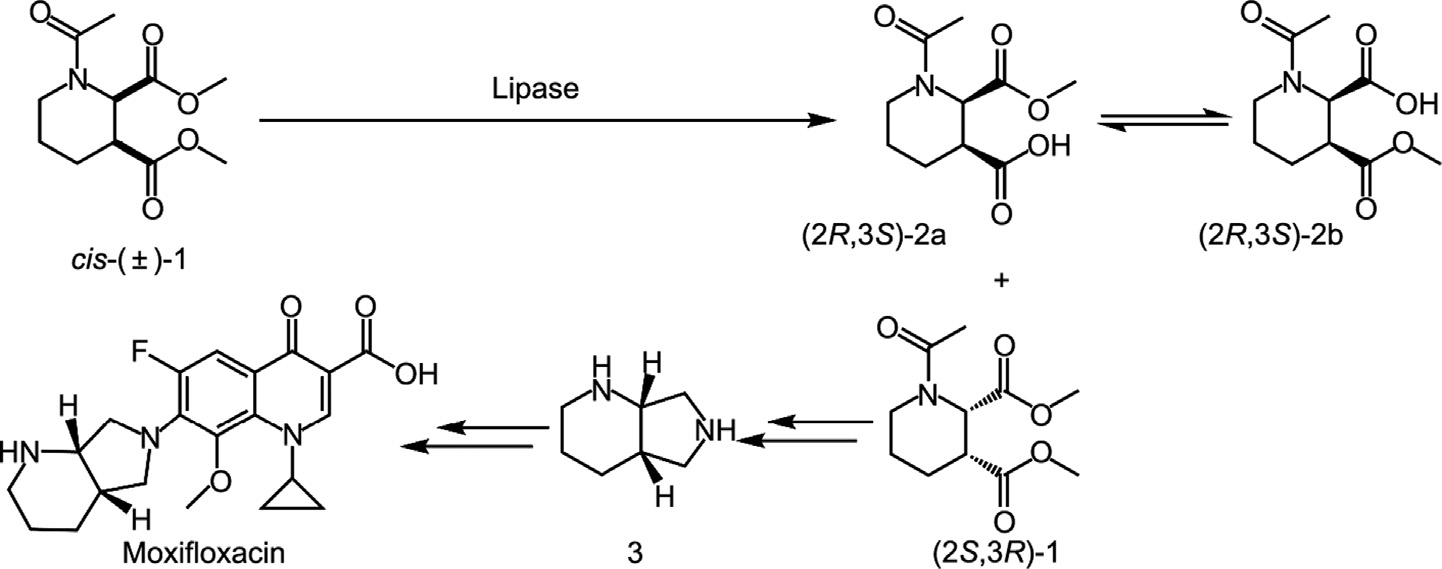
Fig. 1. Reaction scheme for the resolution of cis-(±)-1 by lipase.
Previously, chemical synthetic routes involving the chemical resolution synthesis of chiral side chains with 2,3- pyridinedicarboxylic acid as the raw material, high-pressure hydrogenation, tartaric acid resolution, and other steps; chiral source synthesis using chiral materials as the starting material; and an asymmetric synthesis route, which involved introducing a chiral group into a chiral reagent, were reported [2,7,8]. However, these approaches are associated with common problems, such as low efficiency, expensive starting materials, and the use of metal catalysts. The lipase resolution method for producing (2S,3R)- dimethyl 1-acetylpiperidine-2,3-dicarboxylate ((2S,3R)-1) has high atom economy, high stereoselectivity, high optical purity, and does not require a chemical resolution agent compared with the chemical resolution method [2]. Ramesh et al. [2] reported that 80 g∙L–1 of cis-(±)-1 can be completely resolved with 40 g∙L–1 liquid CALB (Addzyme® CALB, 5000 TBU, 1 TBU = 1 μmol butyric acid released per minute per gram of immobilized enzyme at 40 °C and pH 7.5) within 16 h. Recently, a resolution with higher substrate concentration, fewer enzyme usage, and fewer reaction time by a CALB mutant (I189K) was reported by Shen et al. [9]. According to these researchers, 1 mol∙L–1 (243.26 g∙L–1 ) cis-(±)-1 was completely resolved by 0.1 g∙L–1 enzyme in 5 h, with an activity of 34.28 U∙mg–1 , the highest enzyme activity ever reported. A higher substrate concentration of 2 mol∙L–1 (486.52 g∙L–1 ) extended the resolution time to 8 h with 8-fold the enzyme loading compared to 1 mol∙L–1 substrate. The half-life of CALB-I189K at 35 °C was 58 h and was found to completely lose activity at 50 °C for 24 h [9]. The instability and substrate inhibition of the CALB mutants, which are intolerant to harsh industrial conditions, are the limitations of this reaction.
Protein engineering can be divided into three main categories: directed evolution, semi-rational design, and rational design [10,11]. The combination of directed evolution and rational design constitutes the semi-rational design, which is an effective approach that aims to generate ‘‘small but smart” mutant libraries [12,13]. Rational design is primarily based on an in-depth understanding of the molecular structure of enzymes, for which knowledge of the crystal structure of the protein is necessary [14]. More than one thousand lipase structures that are conducive to the rational design of a lipase without a crystal structure, such as a lipase from Sporisorium reilianum (SRL) identified to possess excellent thermostability and pH stability (SRL’s half-life time at 60 °C was 238 h, retaining over 75% at pH 3–11 for 72 h) in our previous work, have been analyzed [15]. Further, many attempts have been made with CALB to obtain mutants with superior industrial process properties by protein engineering, especially on the lid structure, which is at the active site of enzymes responsible for substrate binding. Notably, most prior studies focused on improving the stereoselectivity and stability of lipases for various substrates [16–19]. Wild-type SRL displayed remarkable stability and stereoselectivity toward cis-(±)-1; however, the very low activity of cis-(±)-1 is a limiting factor. Therefore, a combinatorial approach (rational design and site-directed mutagenesis) was employed for wild-type SRL tailoring to obtain enzyme activityimproved variants that are suitable for the industrial production of moxifloxacin intermediates.
《2. Material and methods》
2. Material and methods
《2.1. Plasmids, strains, and chemicals reagents》
2.1. Plasmids, strains, and chemicals reagents
The recombinant plasmids, pET22b-SRL and pPICZα-A-SRL, were constructed as previously described [15]. Escherichia coli (E. coli) DH5α (Invitrogen, USA) was used for cloning, and E. coli Rosetta (DE3) and Pichia pastoris X-33 (Novagen, Germany) were used to express SRL and its variants, respectively. ZeocinTM, p-nitrophenol, and p-nitrophenyl (p-NP) esters (C2–C16) were purchased from Aladdin Industrial Corporation (China). All other chemicals were of analytical grade and were commercially available.
《2.2. Cell culture and expression》
2.2. Cell culture and expression
E. coli Rosetta (DE3)/pET22b-SRL cells were cultivated in 96- well F-bottom microtiter plates. Luria–Bertani (LB) medium (5 g∙L–1 yeast extract, 10 g∙L–1 tryptone, and 10 g∙L–1 NaCl) containing 100 mg∙L–1 ampicillin and 20 mg∙L–1 chloramphenicol was inoculated with 5 μL of pre-culture and cultivated at 37 °C for 5 h. Isopropyl β-D-thiogalactoside was then added to a final concentration of 0.1 mmol∙L–1 to induce SRL expression, and the cells were cultivated at 22 °C for 12 h before harvest via centrifugation (Eppendorf 5810R; 4 °C, 6000g (g: gravitational acceleration), 15 min). Plasmid pPICZα-A-SRL expressed in the Pichia pastoris X-33 strain was cultivated in yeast extract–peptone–dextrose (YPD) medium (10 g∙L–1 yeast extract, 20 g∙L–1 tryptone, and 20 g∙L–1 glucose, pH 7.0) containing Zeocin 100 μg∙mL–1 . The cells were grown at 30 °C for three days, with methanol (1% v/v) addition every 12 h for induction. The protein expression of SRL was assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
《2.3. Homology modeling and structural analysis》
2.3. Homology modeling and structural analysis
The tertiary structures of SRL were predicted using SWISSMODEL (https://swissmodel. expasy. org/) [20], with CALB (Protein Data Bank (PDB): 1TCA) as a template, and validated using the PROCHECK program. Molecular docking was performed using AutoDock 4.2 with a genetic algorithm to investigate the molecular interactions [21]. Salt bridges within 4 Å were predicted using VMD 1.8.6 [22]. Molecular docking was also performed using AutoDock Vina (https://vina.scripps.edu/) [23] with a genetic algorithm to investigate the molecular interactions between the SRL variants and ligands. All images of the molecular model were generated using PyMol 2.1 (https://www.pymol. org).
《2.4. High-throughput screening system for the detection of highactivity SRL variants》
2.4. High-throughput screening system for the detection of highactivity SRL variants
High-throughput screening of SRL variants was performed as previously described [9]. Briefly, harvested cells in 96-well microplates were resuspended in 400 μL of 5 mmol∙L–1 Tris–HCl buffer (pH 8.5). The hydrolytic reaction was conducted in 5 mmol∙L–1 Tris–HCl buffer at pH 8.5. Each well was added with 100 μL substrate solution (100 mmol∙L–1 calcium chloride and 20 g∙L–1 dimethyl 1-acetylpiperidine-2R,3S-dicarboxylate (2R,3S)-1 or dimethyl 1-acetylpiperidine-2S,3R-dicarboxylate (2S,3R)-1), 20 μL indicator (0.5 mg∙mL–1 bromothymol blue solution), and 100 μL cell suspension sequentially. Thereafter, the plates were incubated at 37 °C. Variants with enhanced activity and high enantioselectivity were detected by color change and further confirmed by highperformance liquid chromatography (HPLC) analysis.
《2.5. Site-saturation mutagenesis and combinatorial mutagenesis》
2.5. Site-saturation mutagenesis and combinatorial mutagenesis
Site-saturation mutagenesis (SSM) libraries of SRL at positions 145, 149, 154, 156, and 159 were constructed using a one-step error-prone polymerase chain reaction (PCR) method with the primers listed in Table S1 in Appendix A. The pPICZα-A-SRL plasmid was used as the template. The PCR mix (50 μL) consisted of Pfu Turbo DNA polymerase (2.5 U; Agilent Technologies, USA), deoxyribonucleotide (dNTP) mix (10 mmol∙L–1 ; Agilent Technologies), template (10 ng), and double-distilled water. The following cycling conditions were employed for PCR: 98 °C for 1 min, one cycle; 98 °C for 30 s, 61 °C for 30 s, 72 °C for 7 min, 3 cycles; 98 °C for 30 s, 61 °C for 30 s, 72 °C for 5 min, 15 cycles; and 72 °C, 5 min, one cycle. After DpnI digestion and purification, the amplified PCR products were cloned into the pET22b(+) vector and transformed into E. coli Rosetta (DE3) cells. The combination of the beneficial substitutions identified by SSM was performed using sitedirected mutagenesis. The sequences of the constructed variant genes were verified by DNA sequencing.
《2.6. Purification of the SRL variants via His-tag affinity》
2.6. Purification of the SRL variants via His-tag affinity
SRL and its variants were cultivated in Erlenmeyer flasks (2 L) using 400 mL of YPD medium containing 100 μg∙mL–1 Zeocin. The SRL fraction was separated by centrifugation (10 000g, 4 °C, 20 min), concentrated, and further purified with a Bio-Scale Mini Nuvia IMAC Ni2+-charged cartridge (5 mL) column (GE Healthcare, Germany). The column was pre-equilibrated with buffer A (50 mmol∙L–1 sodium phosphate buffer containing 0.3 mol∙L–1 NaCl, pH 8.0), non-His-tagged proteins were removed via washing with buffer B (buffer A with 30 mmol∙L–1 imidazole, pH 8.0), and the His-tagged proteins were eluted with buffer C (buffer A with 500 mmol∙L–1 imidazole). The enzyme fraction was dialyzed against 20 mmol∙L–1 sodium phosphate buffer (pH 7.0) and stored at –80 °C. The purity of the enzyme was determined using SDSPAGE, and the concentration of the purified enzyme was estimated using a bicinchoninic acid (BCA) protein assay kit (KeyGen Biotech, China).
《2.7. Standard enzyme activity assay》
2.7. Standard enzyme activity assay
An enzyme activity assay was conducted by measuring the conversion of 50 mmol∙L–1 cis-(±)-1 to (2R,3S)-1. The reaction system (1 mL) comprised 100 mmol∙L–1 sodium phosphate buffer (pH 7.0), 50 mmol∙L–1 cis-(±)-1, and 10 μg of pure enzyme. The reaction mixture was incubated at 35 °C for 30 min and the reaction was terminated by adding 30 μL of 6 mol∙L–1 HCl. The product was extracted with 1 mL of ethyl acetate, and 200 μL of the organic phase was evaporated at 25 °C. Mobile phase of 1 mL was dissolved, and a certain amount of anhydrous sodium sulfate was added to dry the solution. The samples were then subjected to HPLC analysis to evaluate their conversions and stereoselectivities. One unit of enzyme activity (U) was defined as the amount of enzyme required to hydrolyze 1 μmol of (2R,3S)-1 per minute at pH 7.0 and 35 °C.
《2.8. Characterization of SRL and its variants》
2.8. Characterization of SRL and its variants
(1) Effects of temperature and pH on enzyme activity and stability. To determine the pH and temperature profiles, the activities of SRL and its variants were measured in the pH range of 5.0–10.0 and temperature range of 35–65 °C. All buffers used for the pH profile had concentrations of 100 mmol∙L–1 . The following buffers were used: sodium citrate (pH 5.0, 5.5, and 6.0), sodium phosphate (pH 6.0, 6.5, 7.0, 7.5, and 8.0), Tris–HCl (pH 8.0, 8.5, and 9.0), and sodium bicarbonate (pH 9.0, 9.5, and 10.0). The activity was determined at 35 °C. For pH stability measurement, the purified enzymes were incubated in different buffers (100 mmol∙L–1 , pH 5.0–10.0) at 35 °C for 3 h before detecting the residual activities. The temperature profile was determined using a sodium phosphate buffer (100 mmol∙L–1 , pH 7.0). In addition, the temperature stability was analyzed. Therefore, the enzyme solutions were incubated at 50 °C for 0–96 h and the residual enzyme activities were measured at 35 °C. The temperature stability of the best beneficial variant of SRL at 60 °C was also determined.
(2) Kinetics parameters determination. The kinetic constants of the purified enzymes on the substrate cis-(±)-1 were determined by measuring the initial activity at various substrate concentrations (2–200 mmol∙L–1 ). The values and corresponding errors for the maximum velocity (Vmax) and the Michaelis constant (Km) were calculated from the Michaelis–Menten equation by non-linear regression fitting.
(3) Stability determination by circular dichroism (CD) spectrometry. The effects of temperature on the stability of SRL and its variants were further investigated using a ChirascanTM CD spectrometer (Applied Photophysics Ltd., UK) equipped with a TC-1 temperature controller (Quantum Northwest, China) to determine changes in the secondary structure and the melting temperature (Tm) values. Sodium phosphate buffer (20 mm, pH 7.0) at a concentration of 0.1 mg∙mL–1 purified enzyme, temperatures ranging from 30 to 90 °C, a 1 mm pathlength cell, and a temperature rising rate of 1 °C∙min–1 were employed. CD spectral data were collected in the wavelength range of 180–260 nm. The data were processed and analyzed using Global 3 analysis software.
《2.9. Product quantitation via HPLC》
2.9. Product quantitation via HPLC
HPLC analysis was performed as previously described [15] using a Shimadzu UFLC system (Shimadzu Co., Ltd., Japan) coupled with an SPD-20A UV detector. Chiralpak® IG (Φ4.6 mm × 250 mm, 5 μm; Daicel, Japan) was employed as the column, and a mixture of 60% n-hexane and 40% ethanol was employed as the mobile phase at a flow rate of 1.0 mL∙min–1 at 220 nm. The column temperature was set at 30 °C, the injection volume for each sample was 10 μL, and the retention times were 11.6 min for (2R,3S)-1 and 14.7 min for (2S,3R)-1. All samples were measured twice.
《2.10. Enzymatic resolution of cis-(±)-1 by the SRL variant》
2.10. Enzymatic resolution of cis-(±)-1 by the SRL variant
To further explore the industrialization potential, enzyme resolutions of cis-(±)-1 were performed in a 30 mL system containing 0.1 g∙L–1 purified SRL variant and concentrations of 1.0, 1.5, and 2.0 mol∙L–1 cis-(±)-1 in 100 mmol∙L–1 sodium phosphate buffer (pH 7.0), with magnetic stirring at 35 °C, respectively. The pH of the reaction was automatically maintained at 7.0 through titration with 2 mol∙L–1 NaOH during the entire process, and samples were taken periodically to measure the conversions.
《2.11. Constrained molecular dynamics simulation》
2.11. Constrained molecular dynamics simulation
Molecular dynamics (MD) simulations of SRL and its variants were performed according to the method described by Cai et al. [12], with minor modifications. Simulations were performed at 323 K for 20 ns using GROMACS 5.1 [24] and the Amber99SBILDN force field [25]. The structures were dissolved in transferable intermolecular potential 3P (TIP3P) water in a cubic box with a minimum distance of 1 nm from the edge of each lipase. A physiological concentration (154 mmol∙L–1 ) of NaCl was added to neutralize the system. After energy minimization and equilibration in terms of temperature and pressure, an integral step size of 0.002 ps was carried out for each system under isothermal– isobaric conditions (35 °C, 1 bar (1 bar = 105 Pa)). To investigate the stability of the proteins, root-mean-square deviation (RMSD) analyses were performed using GROMACS analysis tools.
《2.12. Statistical analysis》
2.12. Statistical analysis
Statistical analyses were performed using Excel 2011 (Microsoft Corporation, USA). Analysis of variance (ANOVA) was performed using SAS program version 8.1 (SAS Institute Inc., USA). All enzymatic assays and analytical measurements were performed at least in triplicate.
《3. Results and discussion》
3. Results and discussion
《3.1. Structural comparison of wild-type SRL and CALB》
3.1. Structural comparison of wild-type SRL and CALB
Protein engineering based on structural analysis is a popular and time-saving approach for improving the thermal stability and catalytic activity of an enzyme; however, the crystal structure is complicated and time-consuming to obtain and analyze [14]. The massive expansion of available protein information in databases, which serve as a free and valuable resource, can be used to meet the demands of researchers in the protein engineering field for protein structure prediction [26]. Initially, a comparative structural study of wild-type SRL and CALB was carried out to gain an understanding of the structure–function relationship in SRL and subsequently guide the protein engineering of SRL for enhanced activity.
The three-dimensional structure of SRL was modelled based on the X-ray crystal structure of CALB (PDB: 1TCA) by Uppenberg et al. [27], where the amino acid sequence identity between SRL and CALB was 73.5%. Sequence alignment results revealed the same catalytic triad (S110, D192, and H230 in SRL), oxygen anion hole (T46 and Q111 in SRL), and three pairs of disulfide bonds (C28– C70, C222–C264, and C299–C317 in SRL) in CALB and SRL (Figs. 2 and 3). SRL contains a typical α/β hydrolase fold, with a central β-sheet composed of nine β-strands surrounded by α-helices. Overall, SRL and CALB share a high similarity in catalytic behavior; however, the obvious differences in key amino acid residues lead to significant discrepancy in catalytic activity, where SRL and CALB had respective activities of 0.04 and 0.12 U∙mg–1 toward cis-(±)-1.
《Fig. 2》
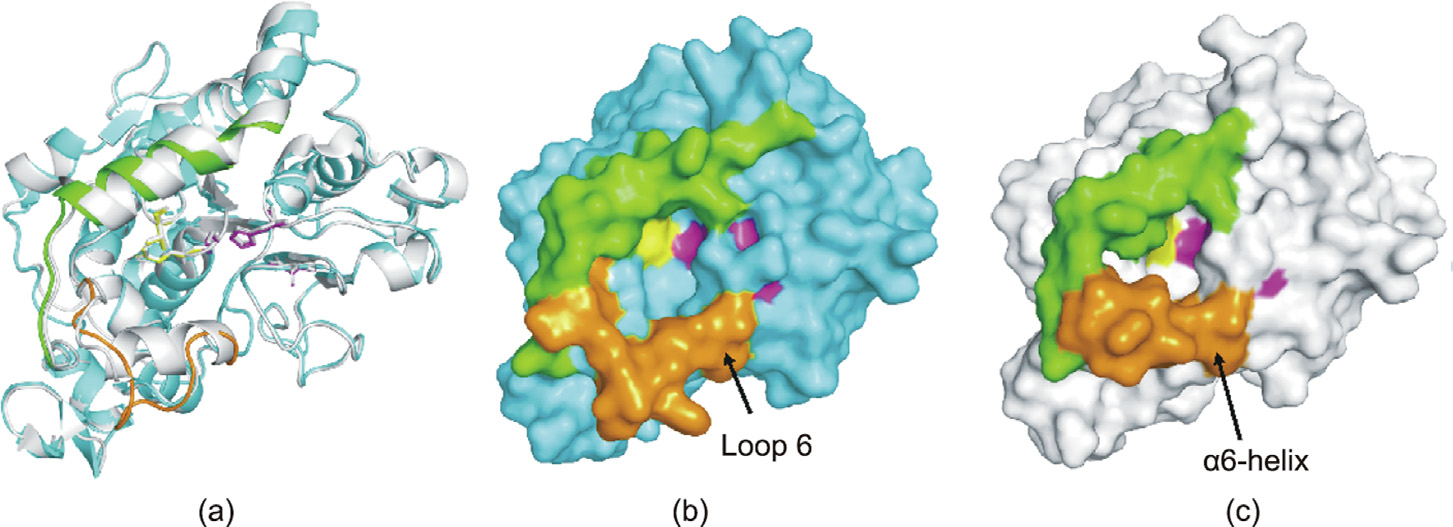
Fig. 2. Three-dimensional model of SRL. (a) Structure overlay of homology-modeled SRL (cyan) and CALB (1TCA, gray). The catalytic triad Ser110–Asp192–His230 in SRL is shown in magenta, the oxyanion hole (T46 and Q111) is shown in yellow, the loop 6 in SRL is shown in orange, and α10 is shown in green. (b) and (c) represent the molecular surfaces of the predicted (b) SRL and (c) CALB, respectively.
As our previous modification of CALB resulted in variants at sites 189 and 190, with the highest activity of a 286-fold increase toward cis-(±)-1 (CALB-I189K) [9], we assumed that mutagenesis on the corresponding sites in SRL (194 and 195) would have similar effects based on the sequence and structural similarities between CALB and SRL. The resulting six variants, namely SRL-I194A, SRLI194R, SRL-I194K, SRL-I194Y/V195L, SRL-I194H/V195L, and SRLI194N/V195L, displayed enhanced catalytic activity and strict stereoselectivity toward cis-(±)-1 compared to the wild-type SRL (Table 1). The optimal variant, SRL-I194N/V195L (V6), had a specific enzyme activity of 10.2 U∙mg–1 , which is 255-fold higher than that of wild-type SRL, but only one-third of that of CALB-I189K (34.28 U∙mg–1 ) [9]. Interestingly, SRL-I194K displayed a slight increase in activity (0.72 U∙mg–1 and 18-fold to wild-type SRL), incomparable to the same residue at position 189 in CALB, which had a 286-fold increase relative to wild-type CALB. Cen et al. [28] reported a highly active cysteine–lipase with a Cys–His–Asp catalytic triad. Further, additional mutations, such as W104V/ A281Y/A282Y/V149G, can evolve, showing a 40-fold higher catalytic efficiency than wild-type CALB in the hydrolysis of 4- nitrophenyl benzoate, and tolerating bulky substrates. Stauch et al. [29] reported two independent protein monomers, A and B, in PDB structure 5A71 of CALB. The residue range, Leu140– Leu147, in monomer B undergoes a remarkable conformational change to form an unfolded loop. At the corresponding region in SRL, the conformation was predicted as a loop, and this domain was located at the entrance of the active pocket, acting as a lid to control the entry of the substrate and release of the product. Accordingly, amino acid residues in loop 6 may have a remarkable impact on SRL catalytic activity, and further protein engineering should be focused on this region.
《Table 1》
Table 1 Specific activities and enantioselectivity (E) of SRL and its variants in the resolution of cis-(±)-1.

《3.2. SSM of SRL based on rational design and combinatorial mutagenesis》
3.2. SSM of SRL based on rational design and combinatorial mutagenesis
Although the residues were narrowed to 14 in loop 6, further shrinking of the mutation library was favored using the online tool, Hotspot Wizard (Fig. S1 in Appendix A) [30]. SSM was performed on residues L145, L149, L154/S156, and V159 using SRL-I194N/ V195L (V6) as the parent. A total of 1600 variants were screened from four constructed saturated mutant libraries with two and three positive variants at the L145 and L154/S156 sites, respectively, which were obtained by primary screening, re-screening, and sequencing confirmation (Table 2 and Fig. S2 in Appendix A). No positive variants were found at sites L149 or V159. Variants V7, V8, V10, and V11 were triple-site variants at L145N, L145V, L154G, and S156H, respectively, whereas V9 was a tetra-site variant at L154G and S156H. By comparing the activity using the colorimetric screening method, the triple-site variant V10 displayed the highest activity, even higher than that of the tetra-site variant V9, indicating the significance of the mutation, L154G. In addition, V7 (L145N) and V8 (L145V) had higher activities than V11 (S156H). Combinatorial variants of L145N and L154G (V12), and L145V and L154G (V13) were constructed based on the parent, V6 (SRL-I194N/ V195L). Variants V7–V13 were used for subsequent enzyme characterization.
《Fig. 3》

Fig. 3. Amino acid sequence alignment between CALB and wild-type SRL (SRL_WT). The secondary structure elements and the residues are depicted based on the previously reported structure of CALB (PDB: 1TCA; CALB_1TCA). The catalytic triad (S110, D192, and H230 in SRL) are indicated by a red star, the oxygen anion hole (T46 and Q111 in SRL) is indicated by a green circle, and three pairs of disulfide bonds (C28–C70, C222–C264, and C299–C317 in SRL) are numbered 1, 2, and 3, respectively. Residues in loop 6 (144–157) and α10-helix (283–293) in SRL are enclosed in a blue box. Residues with SSM in SRL (L145, L149, L154, S156, and V159) are indicated by blue triangles. The alignment was performed using ESPript.
《Table 2》
Table 2 Specific activities, enantioselectivity, and kinetics parameters of the purified wild-type SRL (V0) and variants in the resolution of cis-(±)-1.

Spec EA: specific activity; Rel EA: relative activity; kcat: turnover number; ND: not detected.
《3.3. Characterization of SRL and its variants》
3.3. Characterization of SRL and its variants
3.3.1. Enzyme activity
The hydrolytic activity and stereoselectivity toward cis-(±)-1 were determined by HPLC analysis using purified wild-type SRL (V0) and eight variants (V6, V7–V13) (Figs. S3 and S4 in Appendix A). The specific enzyme activities of the triple-site variants, V7 (SRL-I194N/V195L/L145N) and V8 (SRL-I194N/V195L/L145V), were 21.5 and 27.0 U∙mg–1 , respectively, which were 2.1- and 2.6-fold higher than that of the parent, V6 (SRL-I194N/V195L) (Table 2). V10 (I194N/V195L/L154G) had the highest activity of the triplesite variant, with a value of 30.3 U∙mg–1 , which is 3-fold higher than that of V6, and 757-fold higher than that of V0. To the best of our knowledge, the tetra-site variant, V13 (SRL- I194N/V195L/ L145V/L154G), was found to display the highest enzyme activity of 87.8 U∙mg–1 that has ever been reported toward cis-(±)-1; this activity is 8.6-fold higher than that of V6, 2195-fold higher than that of V0, and 2.6-fold higher than that of CALB-I189K (34.3 U∙mg–1 ) [9]. Variant V11 (S156H) had activity similar to that of parent V6, whereas V9 with an additional mutation of L154G had a 2.5-fold higher activity than V11. Based on a comparison of the enzyme activities of the tetra-site variant (V13) and triple-site variant (V8), V13 displayed a 3.3-fold increase in activity compared with V8. Further comparisons between V11 and V9, and V13 and V8 revealed residue importance at site 154. All SRL variants maintained strict stereoselectivity toward cis-(±)-1, with an enantioselectivity of more than 200.
3.3.2. Kinetics parameters
The kinetic parameters of the SRL variants were determined, and are listed in Table 2. As the enzyme activity of wild-type SRL was very low when cis-(±)-1 was used as the substrate, the kinetic parameters of wild-type SRL were difficult to measure accurately. The affinity of the SRL variants to substrate cis-(±)-1 markedly differed from that of wild-type SRL, in relation to the mutation of amino acid residues at sites 194 and 195. These two sites are located in the binding pocket that interacts with the substrate. The decreased Km value of V13 indicated that variant V13 was most favored for enzymatic catalysis using the substrate cis-(±)-1 after mutagenesis. The Vmax number of V13 (SRL-I194N/V195L/L145V/ L154G) increased significantly from 63.0 μmol∙(mg∙min)–1 of parent V6 (SRL-I194N/V195L) to 238.4 μmol∙(mg∙min)–1 , and the turnover number kcat value of V13 was 3.8-fold higher than that of V6. The kcat/Km value of the optimal variant V13 was 15.4-fold higher than that of the parent (V6) and 1.4-fold higher than that of the CALB optimal variant, CALB-I189K [9]. Variants V9 and V11 contained an S156 mutation that did not have a significant effect on the enhancement of enzyme activity; thus, no further experiments were conducted on V9 and V11.
3.3.3. Temperature and pH effects
Optimal temperature and pH shifts are often observed due to the overall charge variation on the surface of variants [9,12]; thus, the temperature and pH effects on the activities and stabilities of SRL variants must be determined in different pH buffers (pH 5– 10) at 35 °C. The optimal pH values of all SRL variants shifted from 5.5 to 7.0 (Fig. 4(a)). Many microbial lipases are thermostable (optimally active at temperatures higher than 70 °C) and alkali stable (optimally active at pH of greater than 9) [31], and SRL and its variants showed specific acid stability. Based on the CALB variants, the optimal pH shifted to an alkaline environment after protein engineering at the binding pocket [9]. The introduction of the polar amino acid, asparagine at site 194 and leucine at site 195, may change the electrostatic potential near the active pocket. Except variant V6 (SRL-I194N/V195L), other variants had a wider optimal pH range than wild-type SRL; typically, V13 (SRL-I194N/ V195L/L145V/L154G) retained more than 60% of its relative activity ranging from pH 5.0 to 9.0 (Fig. 4(a)). Excellent pH stability was also observed for all variants (Fig. 4(b)). More than 90% of the initial enzyme activities were retained in SRL and its variants incubated at 35 °C for 6 h in the pH range of 4.0–10.0. These results suggest that the introduction of mutations at sites 194, 195, 145, and 154 is irrelevant to the high pH stability of SRL.
The optimal temperatures of all variants were consistent with those of wild-type SRL at 55 °C (Fig. 4(c)). When the reaction temperature increased to 65 °C, the catalytic activity of all, except V12 (I194N/V195L/L145N/L154G), decreased significantly. In terms of temperature stability, the values of all variants decreased in contrast to those of the wild-type SRL at different degrees. Wild-type SRL retained 90% of its initial activity after 96 h at 50 °C and pH 7.0, and its half-life was 597.5 h. Only 40% of the half-life value was detected for the double-site variant V6 (t1/2,50°C = 249.3 h). The remaining variants with the introduction of more amino acid mutations had an even lower half-life of 91.8 h (V7), 115.1 h (V8), 110.2 h (V10), and 92.5 h (V13) (Fig. 4(d)). In addition, the stability of variant V13 (four mutations) at 60 °C decreased significantly, retaining only 23% of its initial activity after 9 h of incubation and a half-life value of 4.1 h (Fig. S5 in Appendix A). Compared to the CALB variant, I189R, which displayed a decrease in optimal reaction temperature from 45 to 40 °C [9], variants of SRL retained the same temperature as wild-type SRL. Overall, the greater the introduction of mutations in an enzyme, the greater the enhancement of the catalytic activity; however, this enhancement has negative impacts on the thermal stability.
《Fig. 4》
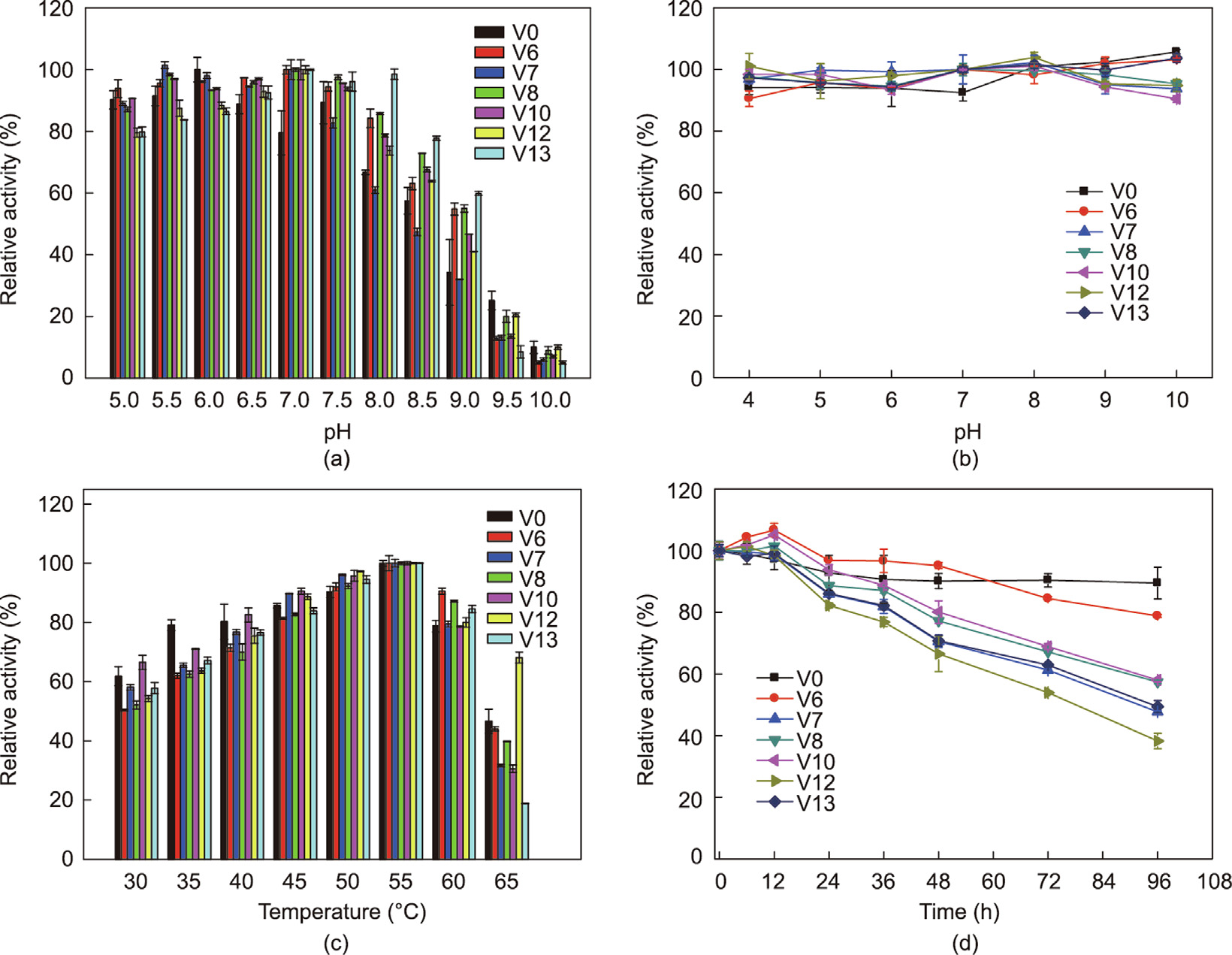
Fig. 4. Effects of pH and temperature on the enzyme activity and stability of SRL (V0) and its variants (V6, V7, V8, V10, V12, V13). (a) Effects of pH on enzyme activity; (b) pH stabilities of wild-type SRL and mutants; (c) effects of temperatures on enzyme activity; (d) thermostabilities of wild-type SRL and variants at 50 °C.
3.3.4. Thermostability analysis based on CD spectrum
To further evaluate the effect of mutation on the thermodynamic stability of SRL and its variants, CD spectrum was used to analyze SRL and its variants (Fig. 5). The positive shoulder band near 192 nm and two negative shoulder bands at 222 and 208 nm are characteristic shoulder bands of the α-helix structure in proteins [32]. The α-helix structure of wild-type SRL did not change significantly at 30–60 °C, whereas the structures of all variants changed and the α-helix structure decreased significantly as temperature increased, typically at 60 °C (Fig. 5(d)). The Tm value of wild-type SRL was 74.3 °C, while that of V6 (SRL-I194N/V195L) decreased to 63.5 °C by 10 °C (Fig. 6). This result was consistent with the thermodynamic stability of V6, confirming that the introduction of mutations at sites 194 and 195 markedly affected the thermodynamic stability of all variants. More introduction of mutations at sites 145 and 154 (V7, V8, V10, V12, and V13) caused a decrease of about 2 °C in the Tm values in contrast to V6; however, the triple-site (V7, V8, V10) and tetra-site (V12, V13) variants had quite similar Tm values.
《Fig. 5》

Fig. 5. CD spectrum of wild-type SRL and its variants at (a) 30 °C, (b) 40 °C, (c) 50 °C, and (d) 60 °C.
《Fig. 6》

Fig. 6. Thermal unfolding curves of wild-type SRL and variants.
《3.4. Enzymatic resolution of cis-(±)-1 by wild-type SRL and tetra-site variant V13》
3.4. Enzymatic resolution of cis-(±)-1 by wild-type SRL and tetra-site variant V13
To investigate the resolution ability of variant V13 (SRL-I194N/ V195L/L145V/L154G), which has the highest enzyme activity toward cis-(±)-1 at high concentrations, enzymatic hydrolysis by V13 at 1.0 mol∙L–1 substrate and higher substrate concentrations up to 2.0 mol∙L–1 was carried out (Fig. 7). The reaction rate of variant V13 at 1.0 mol∙L–1 substrate was fast, leading to a conversion rate of 49.9% after 2 h with E > 200, which is significantly higher than that of CALB-I189K which required 5 h to reach 49.9% [9]. In contrast, the conversion rate of wild-type SRL was only 0.2% after 2h(Fig. 7(a)). According to Xu et al. [16], the amino acid residues at sites 189 and 190 in the CALB binding pocket are significant for stereoselectivity and four variants could synthesize all possible product stereoisomers. The tetra-site variant, V13, not only retained high stereoselectivity similar to the wild-type SRL at high concentrations, but also displayed a remarkable boost in catalytic activity. In addition, the time required to reach a conversion rate of 49.9% at a substrate concentration of 1.5 mol∙L–1 extended to 6 h, which was catalyzed by V13 at the same enzyme loading of 0.1 g∙L–1 at a concentration of 1.0 mol∙L–1 , and the initial rate of hydrolysis at 1.5 mol∙L–1 was basically consistent with the substrate concentration of 1.0 mol∙L–1 (Fig. 7(b)). The hydrolysis process curve for the increased substrate concentration of 2.0 mol∙L–1 revealed that the hydrolysis rate gradually decreased and took a longer time of 20 h to reach a conversion rate of 49.7%. Accordingly, a high substrate concentration was inferred to destroy the structure of the SRL variant V13 to some degree; however, V13 still had a higher catalytic activity and substrate tolerance than CALB-I189K owing to its superior structural stability relative to that of CALB-I189K [9]. The high substrate concentration resolution of V13 with high enzyme activity, high yield, and high stereoselectivity is favorable for the industrial production of (2R,3S)-1, making it a potential catalyst for the resolution of cis- (±)-1.
《Fig. 7》
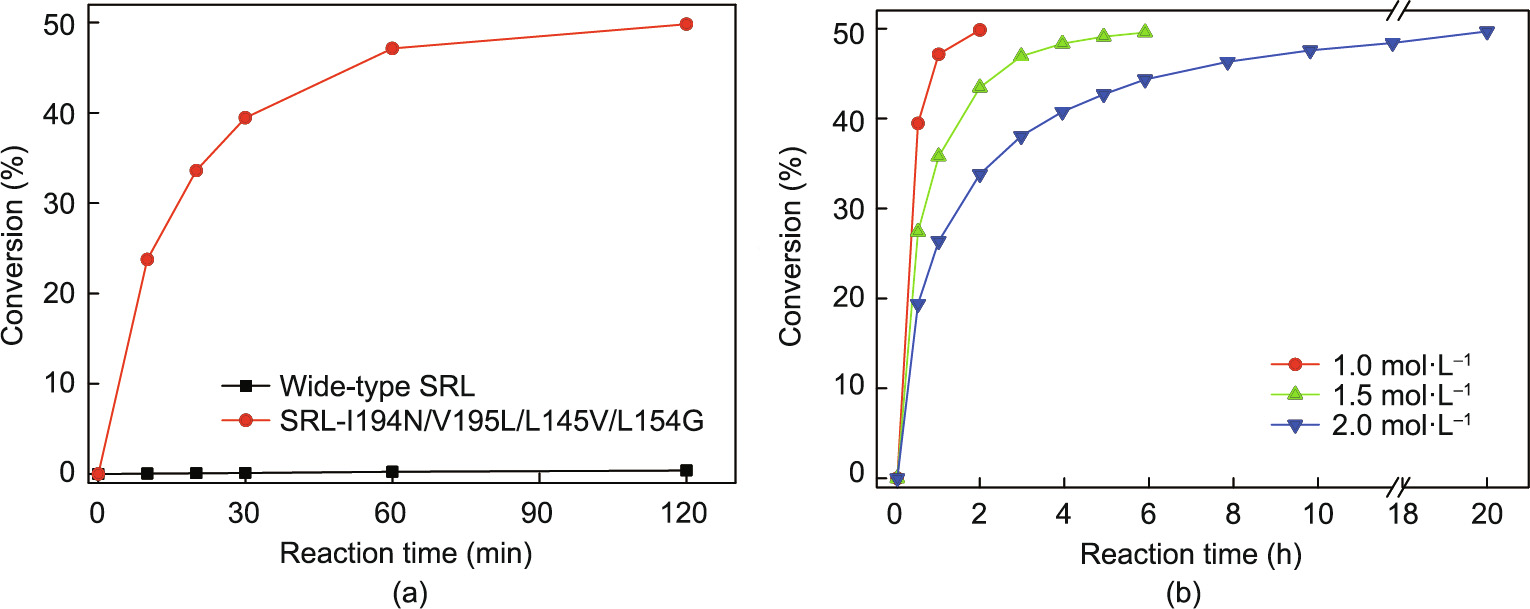
Fig. 7. Enzymatic resolution process curves of the resolution of cis-(±)-1 using purified wild-type SRL and tetra-site variant V13 (SRL-I194N/V195L/L145V/L154G). (a) 1 mol∙L–1 substrate with wild-type SRL (0.1 g∙L–1 ) and V13 (SRL-I194N/V195L/L145V/L154G) (0.1 g∙L–1 ), (b) 1.0–2.0 mol∙L–1 substrate with V13 (SRL-I194N/V195L/L145V/ L154G) (0.1 g∙L–1 ). The reactions were conducted at 35 °C, and the pH was maintained at 7.0 via automatic titration with 2 mol∙L–1 NaOH.
《3.5. Structural analysis of SRL and its variants by MD simulation》
3.5. Structural analysis of SRL and its variants by MD simulation
Three-dimensional structural models of wild-type SRL and its variants, V6, V8, V10, and V13, were constructed. SRL and its variants were found to be in equilibrium after 5 ns of MD simulation based on the RMSD values, which reflected the vibration of the carbon skeleton in the protein structure during the entire simulation process and implied that the enzyme molecules were in a stable conformation in the solvent box for subsequent analysis (Fig. S6 in Appendix A).
The binding sites of product (2R,3S)-1 in the catalytic pocket of wild-type SRL (V0) and its variant, SRL-I194N/V195L (V6), were compared, and the binding position of the product in the catalytic activity pocket was shifted (Fig. 8). The shape of the catalytic pocket of variant V6 differed from that of the wild-type SRL. The catalytic mechanism of CALB has been explained since the 1990s and important progress was made regarding the crystal structure after 2010 [33]. The amino acid residue, isoleucine, at 194 with large steric hindrance and hydrophobicity was mutated to a hydrophilic amino acid residue, asparagine, with smaller steric hindrance, which not only reduced the hindrance to the substrate molecules, but also formed a new hydrogen bond between the acylamino group of N194 and the acyl group of the second ester bond of the substrate, ultimately producing an obvious fixed force on the substrate, making the substrate molecule positioned in a state of easy hydrolysis. The shorter distance r (O–C) between the oxygen atom of serine at site 110 and the acyl carbon atom of the substrate in variant V6 was a possible reason for the improvement in enzyme activity (Fig. 8). Stauch et al. [29] reported similar results for the CALB structure 5A71. The distance reduction was conducive to the formation of a tetrahedral transition state and the nucleophilic attack of an oxygen anion from S110 on substrate (2R,3S)-1. The shorter distance between the substrate acyl oxygen and oxygen anion hole was favored for hydrogen bond formation, stabilized the substrate in the tetrahedral transition state, and was conducive to the catalytic reaction.
《Fig. 8》

Fig. 8. Molecular docking of (2R,3S)-1 (green) into the active site of (a) wild-type SRL and (b) the variant, SRL-I194N/V195L. The catalytic triad, D192–H230–S110, is displayed in red, the oxyanion hole is displayed in blue, the residues in wild-type CALB are indicated in orange (I194, V195), and the mutated residues are indicated in yellow. H bonds for the interaction and stabilization of the oxyanion are indicated by green dotted lines. The distances for effective nucleophilic attack of serine in the catalytic triad at the carbonyl function of (2R,3S)-1 are labeled as yellow dotted lines.
Further enhancement of the catalytic activities of SRL-I194N/ V195L/L145V (V8), SRL-I194N/V195L/L154G (V10), and SRLI194N/V195L/L145V/L154G (V13) may be related to changes in the lid moiety in SRL (Fig. 9). In most lipases, an ‘‘interface activation” occurs during catalysis [34,35]. CALB was exceptional to be considered as lack of ‘‘interface activation” effect due to the small lid segment, which is composed of α5 helix, acting as an esterase that can hydrolyze various water-soluble substrates [36]. Recent studies revealed that the α5 helix lid structure of CALB is very flexible, and remarkable conformational changes have been observed in the catalytic process [37]. Through long-term MD simulation, Luan and Zhou [36] reported that the salt bridge interaction between D145 on loop 6 and R309 on the β8 sheet and the hydrophobic interaction between L144, L147, and W155 on loop 6 could stabilize the lid moiety in an open conformation, whereas the interactions between the hydrophobic residues P143, L144, L147, and V149 on loop 6 and V286 and K290 on the α10 helix allowed the lid to adopt a closed conformation. The hydrophobic interaction between A282 and L278 on the α12 helix and I189 in the loop region enhanced the hydrophobic interaction between the loop 6 and α10 helices. In contrast to CALB, the lid region had a more open conformation in SRL; thus, the formation of a salt bridge with residue R314 on the β8 sheet was difficult. The mutated residues at sites 145 and 154 were assumed to be located on flexible loop 6 with a relatively compact conformation in V13, as leucine was changed to valine and glycine, respectively. Loop 6, together with the residues on the α10 helix, provided a suitable clash for ligand binding.
《Fig. 9》
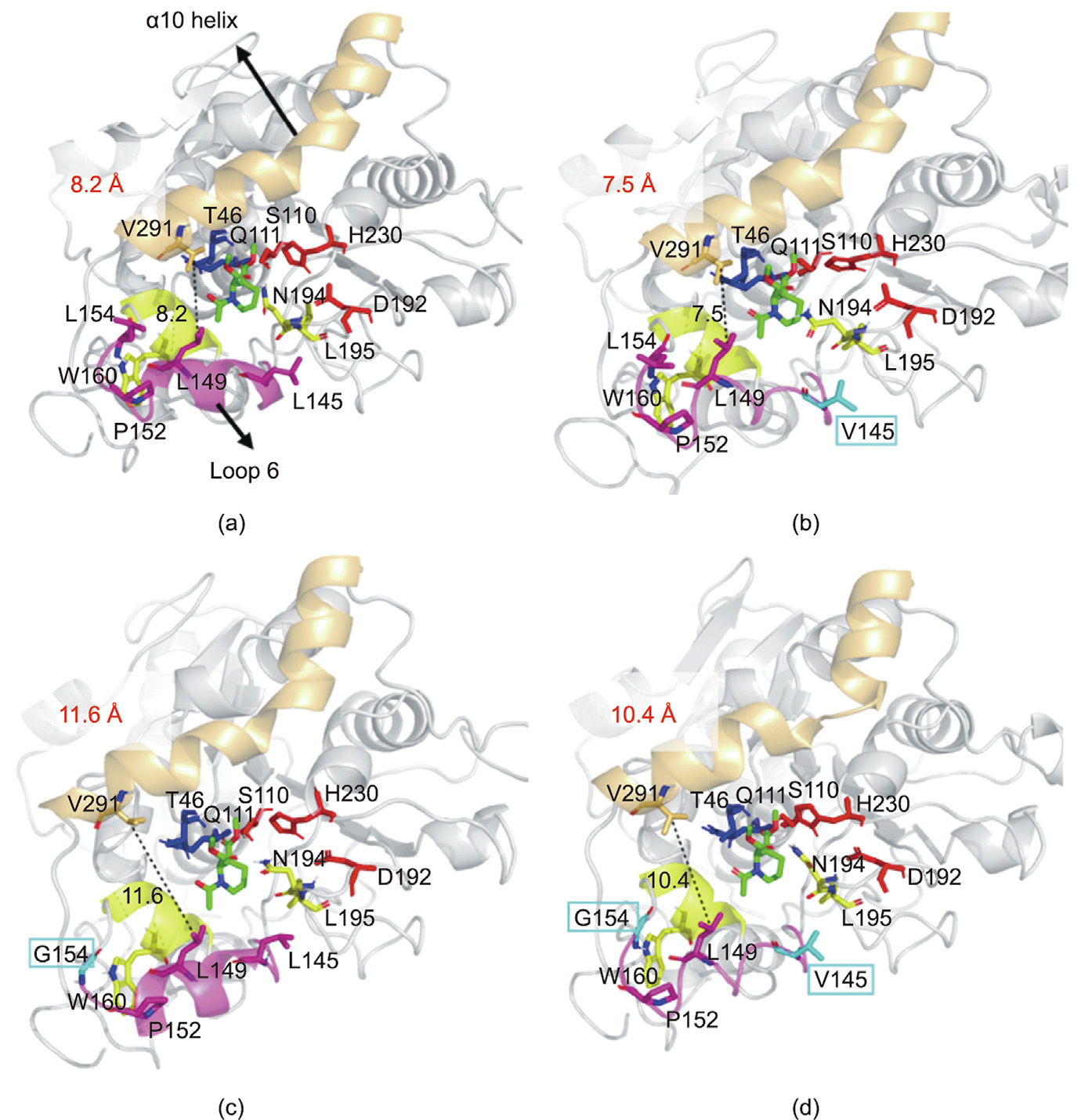
Fig. 9. Molecular docking of (2R,3S)-1 (green) into the active site of the SRL mutants. (a) SRL-I194N/V195L (V6); (b) SRL-I194N/V195L/L145V (V8); (c) SRL-I194N/V195L/ L154G (V10); (d) SRL-I194N/V195L/L145V/L154G (V13). The catalytic triad D192–H230–S110 is displayed in red, the oxyanion hole is displayed in blue, the residues N194 and L195 are indicated in yellow, loop 6 is displayed in purple, the α10 helix is displayed in orange, and the α7 helix is displayed in yellow. The residues in the SRL-I194N/ V195L are colored gray (L145, L154) and the mutated residues are colored cyan. H bonds for interaction and the stabilization of the oxyanion are indicated by grey dotted lines. The distances for V291 and L149 are labeled as black dotted lines.
《4. Conclusions and future prospects》
4. Conclusions and future prospects
SRL was engineered from barely detectable to 87.8 U∙mg–1 for enhanced enzyme activity toward cis-(±)-1 by SSM, site-directed mutagenesis, and combinatorial mutagenesis. Two key amino acid residues at positions 145 and 154 in SRL, relevant to the lid conformation, were identified in the substrate-binding pocket in loop 6, and proper variations of the two key residues resulted in the wide opening of the lid in a manner that enabled significant enhancement of the activity. The findings of this study highlight an enzyme suitable for the industrial process of moxifloxacin production and some key amino acids that are important for enzyme activity. Further work, such as crystallization, will be employed to gain deeper structural insights into SRL and its variants and develop immobilization techniques for enhanced stability, reusability, and convenient separation of products to reach an industrially applicable level.
《Acknowledgments》
Acknowledgments
This work was financially supported by the National Key Research and Development Program of China (2021YFC2102100 and 2018YFA0901400), the Fundamental Research Funds for the Provincial Universities of Zhejiang (RF-C2019005), the National Natural Science Foundation of China (32000898), and the Natural Science Foundation of Zhejiang Province (LQ21B060006).
《Compliance with ethics guidelines》
Compliance with ethics guidelines
Xue Cai, Jiang-Wei Shen, Yu Qiang, Jing Hua, Zhang-Qi Ma, Zhi-Qiang Liu, and Yu-Guo Zheng declare that they have no conflict of interest or financial conflicts to disclose.
《Appendix A. Supplementary data》
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2022.03.020.











 京公网安备 11010502051620号
京公网安备 11010502051620号




