


在文献
氢分子离子是2个质子和1个电子构成的最简单的分子体系, 也是量子力学唯一能获得薛定谔方程精确解的分子体系。尽管H2+结构非常简单, 但其求薛定谔方程精确解已很困难, 即使用简单的原子轨道线性组合 (LCAO) 变分法计算也相当复杂
李政道认为:“世界上一切自然现象, 都是以一组相当简单的自然原理构成基础的”, “常常是问题越基本, 其解越简单。需要的是正确的认识和观念, 而不是繁复的数学和计算”
《1 氢分子离子的结构模型》
1 氢分子离子的结构模型
氢分子离子是在用电子束轰击放电管中的氢气时在光谱中观察到的。它的性质活泼, 寿命很短, 一旦与其他物质接触立即夺取电子而形成氢分子。因此, 笔者认为从化学的角度看不能将其视为一种稳定的分子, 而可以将其视为质子化 (Pronation) 的氢原子, 也即是氢原子束缚了一个裸质子。H2+的产生情况可示意于图1 (及封面) 。
图1中 (a) 表示出按1s电子径向几率密度分布函数的电子云发生交盖形成了氢分子, 氢分子中的2个自旋相反的电子仍然各自围绕着自己的核运动。交盖区中的2个球冠面电子云吸引着a, b两核, 克服了两核间的库仑排斥力, 达到一种巧妙的动态平衡。交盖的程度是交叉点与两核的联线成直角, 因此核间距ab (H2的Re) 的长度为
质子化的原子很多。例如卤化氢离子 (HX+) 就可视为质子化的卤原子, 它们像H2+一样非常活泼, 而且除HF+外, 其余质子化卤原子HCl+、HBr+、HI+的键能比相应分子还略大一些。这可以看作是裸质子与卤原子形成的偶极作用比氢原子与卤原子形成的离子键还要强一些, 见表1。
在化学反应的体系中, 还存在着许多质子化的分子, 例如酸溶液中的H3O+是质子化的水分子;氨溶液中的NH4+是质子化的氨分子。在溶剂萃取反应的有机相中, 醚类、酮类、酯类、胺类等萃取剂分子都非常容易形成质子化的有机分子, 如质子化的TBP (磷酸三丁酯) 其结构式为 (C4H9O) 3P=O∶H+, 质子化的TOA (三辛胺) 其结构式为 (C8H17) 3N:H+, 这两种带正电荷的有机分子均是靠氧或氮原子上的孤对电子束缚了一个质子。
表1 卤化氢分子与相应的分子离子的键能 (kJ·mol-1)
Table 1 The bond-energy of halogenated hydrogen molecule and their corresponding molecule ion (kJ·mol-1)
《表1》
卤化氢分子 | HF | HCl | HBr | HI |
键能 (De) | 566.5 | 431.7 | 366.8 | 298.5 |
分子离子 | HF+ | HCl+ | HBr+ | HI+ |
键能 (De) | 443.4 | 457.6 | 388.1 | 304.4 |
数据引自B.И.Beдeнeeв, Л.В.Гypвич等, 《化学键的离解能, 电离势和电子亲合能》 (俄文版) , 莫斯科, 1962, 42~60。原数据单位为kCal·mol-1, 笔者进行了换算。
此外, 甚至在大量吸氢后的金属钯中, 有人认为PdHx (x<1) 的结构状态是氢原子把它们的价电子传递给金属原子的d轨道, 因而变成可流动的质子。其根据的实验事实是:a.在PdHx中氢的流动性很大;b.当氢的含量增加时, 钯的磁化率下降;c.当一个电场加到PdHx的线状体的两端时, 氢向着负极迁移
《2 氢分子离子结构参数的计算》
2 氢分子离子结构参数的计算
《2.1H2+体系中的吸引作用与排斥作用》
2.1H2+体系中的吸引作用与排斥作用
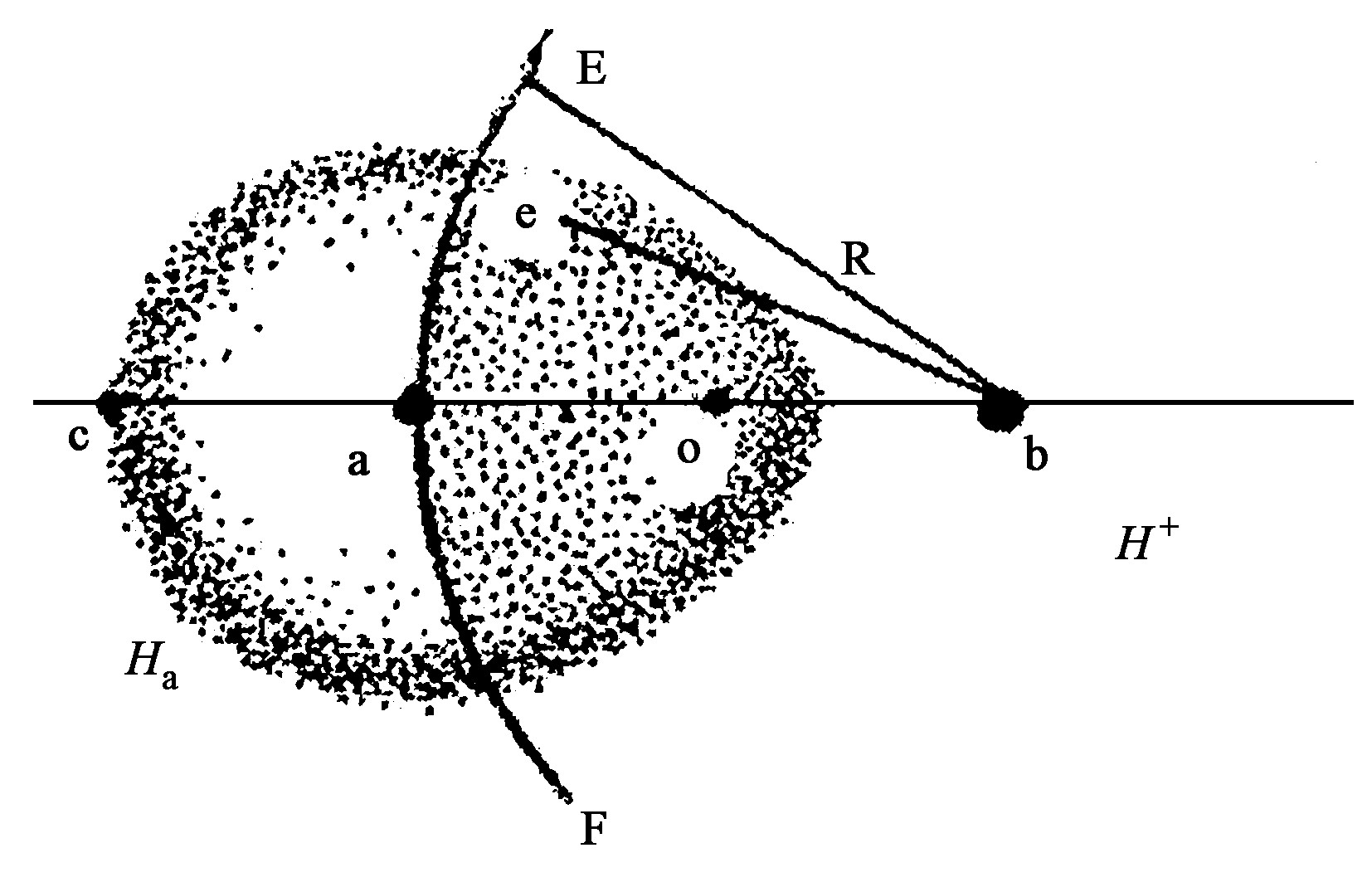
为了计算H2+的结构参数, 先考察一个畸变的氢原子的电子云如何能束缚住一个质子。在图1 (c) 中, 以b为圆心, R=ab为半径作一个球面EF, 如图2:
图2中电子e对a核的吸引作用已为自身的绕核运动所平衡, 可以不作考虑。阴影部分表示以半径为R的球面截出的电子云。使用微观时标如阿秒 (10-18 s) 考察时, 电子e在右边阴影区任何一点出现时, 由于e与b的距离小于ab, 电子对b核的吸引力均将大于两核间的排斥力。当e在ab中点o出现的瞬间, e对a、b两核吸引力之和用简单的式 (1) 很容易算出将大于两核排斥力的8倍。而e在非阴影区, 特别是在c点及其附近出现时, 两核的排斥力最大, 两核距离将因此而拉开。
使用宏观的时间标度 (如秒) 时, e对b核的吸引作用要用电子云考察。显然, 只要畸变后的1s电子云对b核的吸引作用相当于在o点存在一个q=e/8的电荷重心, 则电子云即可束缚住b核。按照笔者的观点
《2.2H2+的平衡核间距Re》
2.2H2+的平衡核间距Re
在H2+中, 由于只有1个电子, 电子云引起两核相向靠近的吸引作用将为H2的1/2。同理, H2+中两核间的排斥力也应只是H2的1/2, 才能达到吸引和排斥的平衡。令Req表示H2的平衡核间距, Re表示H2+的平衡核间距则得下式:
文献
此值与实验值完全一致。
《2.3H2+的键能De》
2.3H2+的键能De
在图1 (c) 及图2中, 当质子b从右边远距离处移近氢原子Ha并形成H2+时, 体系将获得一个数值为
根据以上分析, 则H2+的键能De应为:
将式 (2) 的Re=2a0代入式 (3) 得
使用原子单位时, De=0.109 375 au, 实验值为0.102 530 5 au, 相对误差为6.7%, 比Kolos和Roothaan等
《2.4H2+的振动力常数k》
2.4H2+的振动力常数k
按文献
对于振动力常数还可从另一途径计算。文献
从式 (5) 移项得
用式 (7) 置换去式 (6) 分母中的一个Req, 得下式:
式 (8) 是一个将平衡核间距、键能及力常数关联在一起的公式, 将Req改为H+2的Re, 并将式 (3) 及式 (4) 分别代入式 (8) 则得到:
使用原子单位时, k=0.109 375 au, 这就是说, H2+的键能值和振动力常数完全一致, 仅量纲不同而已。Bishop的De值与k值也非常接近, 笔者的结果与他的一致。
《3 结语》
3 结语
获得的结果列入表2。
Table 2 The equilibrium inter-nuclei distance, bond-energy and vibration force constant of H2+ (au)
《表2》
计算式 | 计算值 | 实验值 | 相对误差/% |
Re=2a0 | 2 | 2 | 0 |
| 0.109375 | 0.1025305 | 6.7 | |
| 0.109375 | 0.102897* | 6.3 |
*采用Bishop的计算值
表2结果表明, 笔者对H2+的平衡核间距、键能及力常数获得了3个十分简洁、物理含义清晰、只含z, e, a0值, 而不含任何人为性参数的计算公式。这些公式不代入Re=2a0时, 与H2的相应公式仅相差一个
像文献











 京公网安备 11010502051620号
京公网安备 11010502051620号




